How GW Pharma Won CBD
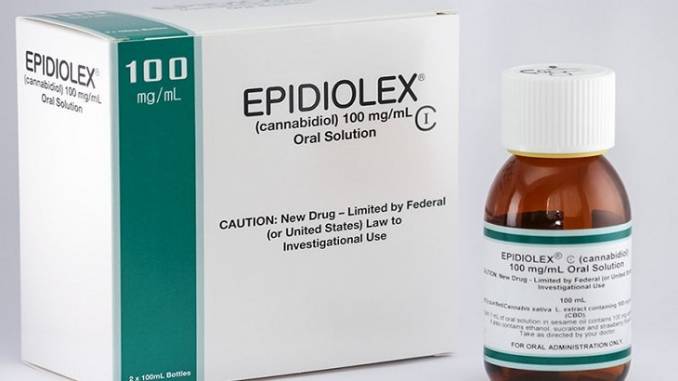

As of this writing, the United States Food and Drug Administration (FDA) has approved GW Pharma’s CBD drug Epidiolex for treating profound refractory pediatric epilepsy syndromes (Dravet syndrome and Lennox Gastaut syndrome) as well as for treating seizures associated with tuberous sclerosis complex (TSC) in patients one year of age or older. The product is a very simple, orally-administered formulation comprised of 100mg/ml cannabidiol (CBD), dehydrated alcohol, sesame seed oil, strawberry flavor and sucralose – basically, an alcohol-based solution with sesame seed oil to help solubilize the CBD oil, flavoring and sweetener.
![]()
It is important to note that the DEA descheduled the Epidiolex formulation and not cannabis-derived CBD itself. Thus, GW Pharma is now in the enviable position of being the only company that can legally sell cannabis-derived CBD. More importantly, because the DEA descheduled the formulation and not the active ingredient, other companies who wish to market cannabis-derived CBD pharmaceutical formulations will have to repeat whatever it is that GW did to get Epidiolex descheduled.3 The DEA effectively gave the company a huge head start with respect to competitors who are developing other cannabis-derived CBD formulations that would compete with Epidiolex. That advantage will remain in place unless and until cannabis-derived CBD itself is descheduled or cannabis is legalized at the federal level.
GW Pharma’s attorneys demonstrated considerable virtuosity in devising this approach. However, there is another aspect of the GW Pharma story – one that could have profound implications for the exploding CBD consumer packaged goods (CPG) industry. The Federal Food, Drug, and Cosmetics Act4 (FFDCA) prohibits the introduction into interstate commerce of any food to which has been added an approved drug or a drug for which substantial clinical investigations have been instituted and made public.5 Because CBD was and is still the subject of clinical trials run by GW Pharma and others, even hemp-derived CBD is currently illegal to use as a food additive or dietary supplement under the FDCA
The FDA has recently re-started the public commentary stage of a long process that will hopefully result in the creation of a regulatory pathway for CBD to be used as a food additive – something that would seemingly be a straightforward matter given the copious amounts of safety data being generated from all of GW Pharma’s clinical trials. However, as long as the FDA continues to drag its feet in providing a regulatory pathway for CBD CPG products, CBD, regardless of its source, will remain illegal to use as a food additive or supplement under either the CSA or the FFDCA despite the existence of safety data obtained through the Epidiolex clinical trials. If, as many people in the industry anticipate, the agency decides to begin enforcement action, this could have a hugely negative impact on the industry.
In addition to the potentially disastrous effect that federal law could have on an important new industry, the federal regulatory scheme introduces unnecessary regulatory complexity and cost by imposing two different regulatory schemes depending on the source of the CBD. CBD derived from hemp is chemically identical to CBD derived from cannabis. Despite that identity, the 2018 Farm Bill nonsensically exempts only hemp-derived CBD from the Controlled Substances Act. If a regulatory pathway is created for hemp-derived CBD, but the DEA insists on maintaining cannabis-derived CBD as a schedule 1 substance, then the same molecule will be subject to two different regulatory schemes. This scenario would require tracking and certifying CBD sources and thereby impose regulatory and economic burdens that are entirely unnecessary from a public health point of view.

Overall, the current state of affairs is unfair, expensive, uncertain and entirely unworkable over the long term. The CSA must be amended, ideally to deschedule both hemp and cannabis entirely, but at least in the short term, to deschedule CBD and preferably all non-THC cannabinoids regardless of their source. Further, the FDA must provide a regulatory pathway to allow the use of low doses of cannabinoids shown to be safe, either by existing clinical trial data or future testing pursuant to the NDIN submission process.
A 2019 Gallup poll found that 14% of Americans – 1 in 7 – use CBD products.7 The demand is there, the industry is thriving, and adequate safety data exists to justify a regulatory system that allows low-dose over the counter CBD products provided those products are produced using Current Good Manufacturing Practices (CGMPs) for food and dietary supplement manufacturing prescribed by the FDA and that such products undergo regular testing that demonstrates they are safe, unadulterated and accurately labeled. It is time for the industry to collectively fund a New Dietary Ingredient Notification (NDIN) submission that would provide safety data sufficiently compelling to force the FDA to either recognize CBD and other non-THC cannabinoids as being GRAS substances regardless of their source, or in the alternative create a regulatory path for CPG products containing low-doses of CBD and other non-THC cannabinoids.
Editor’s Note: The opinions expressed in this publication are those of its author. They do not purport to reflect the opinions or views of the Cannabis Industry Journal, its editorial staff or its employees.
References
- Clincialtrials.gov lists 256 different clinical trials in which Epidiolex has been, is being or will be tested for a wide variety of other indications, including but not limited to opioid use disorder, several types of prostate cancer, alcohol use disorder, musculoskeletal pain, and a host of others.
- REMS – risk evaluation and mitigation strategy – are drug safety programs that the FDA requires in cases where mediations pose serious safety concerns with respect to potential abuse and other adverse effects.
- Exactly what they did isn’t clear, and won’t be for a long while given the snail’s pace at which FOIA requests are filled.
- Title 21 United States Code Chapter 9
- Title 21 United States Cod Chapter 9, Sections 331(ll), 342(a)(1) and Section 342(d)(f)(1)
- “Exclusive: New Report Predicts CBD Market Will Hit $22 Billion by 2022” Rolling Stone Magazine, September 11, 2018, citing cannabis industry analysis from the Brightfield Group.
- Gallup poll on American CBD product usage