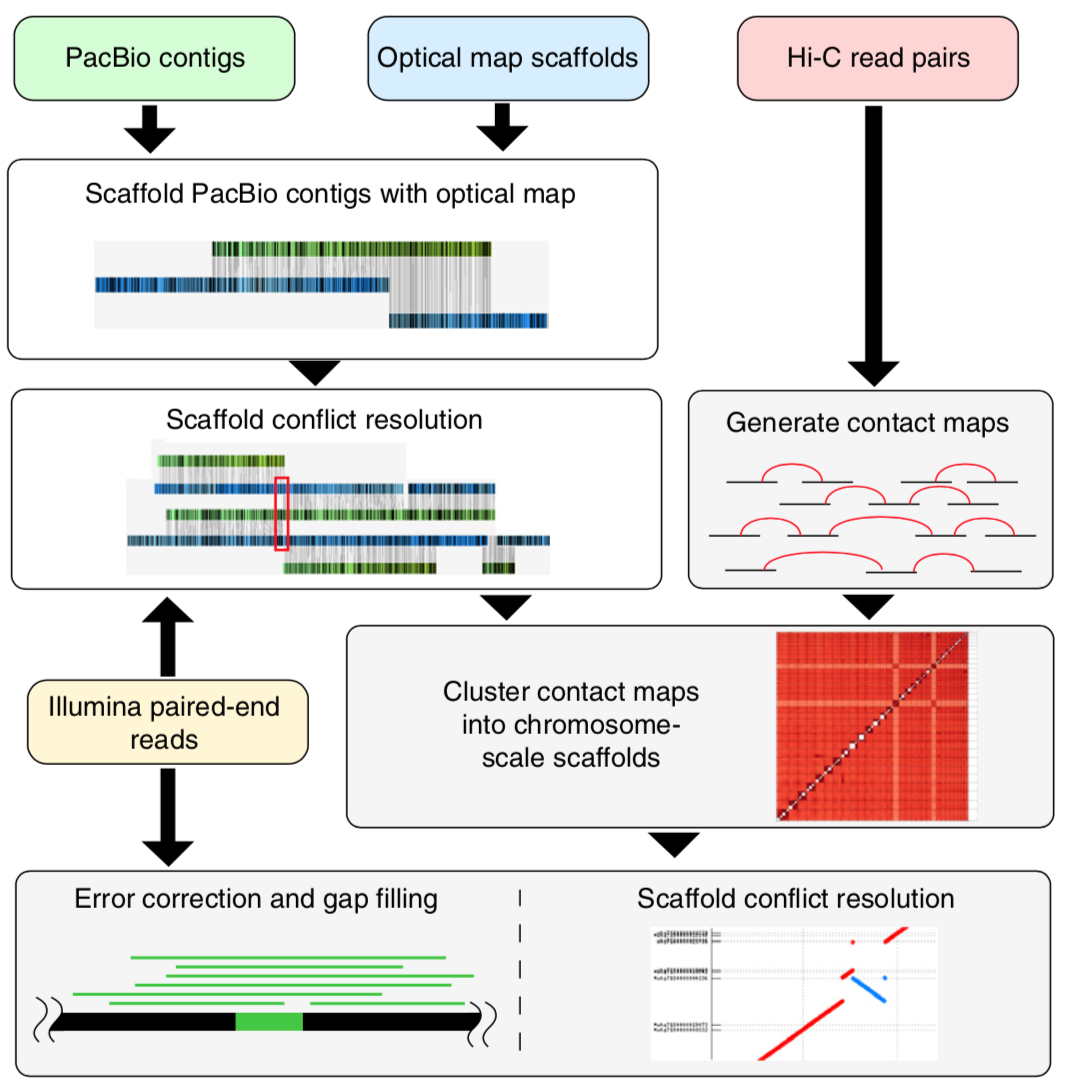
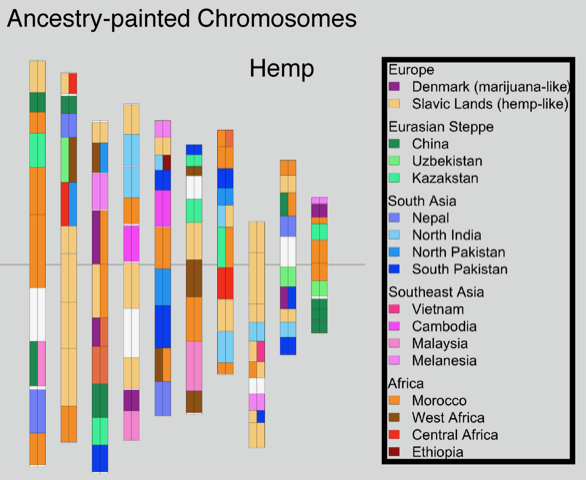
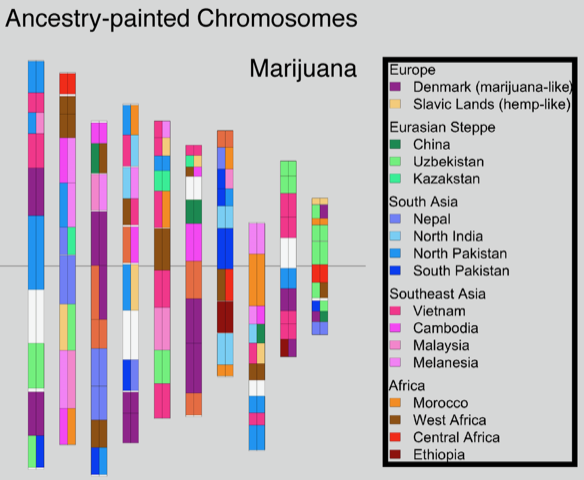
Plant genetics are an important consideration for cultivators planning to grow cannabis crops. Genetics can affect how well a plant grows in a particular environment under various conditions and have a major impact on the production of cannabinoids, terpenes as well as other molecules and traits expressed by the plant.
Front Range Biosciences is a hemp and cannabis genetics platform company, leveraging proprietary next generation breeding and Clean Stock® tissue culture nursery technologies to develop new varieties for a broad range of product applications in the hemp and cannabis industries. FRB has global reach through facilities in Colorado, California and Wisconsin, and a partnership with the Center for Research in Agricultural Genomics in Barcelona, Spain. FRB is headquartered in Lafayette, Colorado.
We spoke with Jonathan Vaught, Ph.D., CEO and co-founder of Front Range Biosciences. Jonathan co-founded Front Range in 2015 after a successful career in the diagnostics and food testing industries.
Aaron Green: Jon, thank you for taking the time today. I saw in the news you recently sent tissue cultures to the International Space Station? I’d love to learn more about that!

Jonathan Vaught: This was a collaborative project between the BioServe group at the University of Colorado Boulder, which is a part of their aerospace engineering program. They do research on the International Space Station, and they have for quite some time. We partnered with them and another company, Space Technology Holdings, a group that’s working on applications of space travel and space research. We teamed up to send tissue culture samples to the space station and let them sit in zero gravity at the space station for about a month, and then go through the reentry process and come back to Earth. We brought them back in the lab to perform some genomic analyses and try to understand if there’s any underlying genetic changes in terms of the plants being in that environment. We wanted to know if there was anything interesting that we could learn by putting these plant stem cells and tissue cultures in an extreme environment to look for stress response, and some other possible changes that might occur to the plants by going through those conditions.
Aaron: That’s an interesting project! Are there any trends that you’re following in the industry?
Jon: We’re excited to see ongoing legalization efforts around the world. We’ve seen continued progress here in the United States. We still have a long way to go, but we’re excited to see the additional markets coming onboard and regulations moving in the right direction. Also, we’re excited to see some of the restorative justice programs that have come out.
Aaron: How did you get involved at Front Range Biosciences?
Jon: It really starts with my background and what I was doing before Front Range Biosciences. I’ve spent more than 15 years developing commercializing technologies in human diagnostics, food safety and now agriculture.

I started my career during graduate school in biotech at the University of Colorado at Boulder, where I helped develop some of the core technology for a human diagnostic startup company called Somalogic here in Colorado. I went to work for them after finishing my dissertation work and spent about six years there helping them grow that company. We ended up building the world’s largest protein biomarker discovery platform primarily serving pharmaceutical companies, hospitals and doctors, with personalized medicine and lab tests for things like early detection of chronic illness, cancer, heart disease and inflammation.
I then went to another startup company called Beacon Biotech, that was interested in food safety. There I helped develop some similar technologies for detecting food-borne illness — things like salmonella, listeria and E. coli. That was my introduction to big food and big agriculture. From there, I went to help start another company called Velocity Science that was also in the human diagnostic space.
Along the way, I started a 501(c)3 nonprofit called Mountain Flower Goat Dairy, a dairy and educational non-profit that had a community milk-share, which included summer camps and workshops for people to learn about local and sustainable agriculture. I became more and more interested in agriculture and decided to take my career in that path and that’s really what set me up to start Front Range Biosciences.
Aaron: Do you have any co-founders?
Jon: I have two other co-founders. They both played various roles over the last four years. One was another scientist, Chris Zalewski, PhD. He currently works in the R&D department and helps oversee several different parts of the company including pathology and product development. My other co-founder, Nick Hofmeister served as chief strategic officer for the last few years, and has helped raise the majority of our funding. We’ve raised over $45 million dollars, and he played a big role in that.
Aaron: What makes you different from other cannabis seed companies?
John: We’ve built the first true cannabis genetics platform. What I mean by that is we built a platform that allows us to develop and produce new plant varieties that support both the hemp and the cannabis markets. To us, it’s all cannabis. Hemp and cannabis are scientifically the same plant. They just have different regulatory environments, different products and different markets, but we stay focused on the plant. Our platform is built on several different pillars. Genetics are one of the core pieces, and by genetics I mean, everything from molecular based breeding to marker assisted breeding to large germplasm collections. We collect different varieties of germplasm, or seed, from all over the world and use those to mix and match and breed for specific traits. We also have large nursery programs. Another one of our pillars of the platform includes greenhouse nursery production — everything from flowering cannabis plants to producing cannabis seeds to cloning and producing mother plants and rooted cuttings or clones.
![]() Then tissue culture is another part of the platform, it’s basically the laboratory version of a greenhouse nursery. It’s housed in a sterile environment and allows us to produce plants that are clean and healthy. It’s a much more effective, modern way to manage the nursery. It’s part of our clean stock program, where we start clean, stay clean, and you can finish clean. It’s really built on all of those different pieces.
Then tissue culture is another part of the platform, it’s basically the laboratory version of a greenhouse nursery. It’s housed in a sterile environment and allows us to produce plants that are clean and healthy. It’s a much more effective, modern way to manage the nursery. It’s part of our clean stock program, where we start clean, stay clean, and you can finish clean. It’s really built on all of those different pieces.
We also have capabilities in analytical chemistry and pathology, that allow us to better understand what drives performance and the plants, and both different regions as well as different cannabinoid products or terpene products. All of the science and capabilities of the platform are what allow us to create new varieties faster, better, stronger.
Aaron: It sounds like you’re vertically integrated on the front-end of cannabis cultivation.
Jon: Absolutely, that’s a great way to think about it.
The last piece I’d say is that we have areas of research and development that cover the full span of multiple product lines. We think about it from an ingredient perspective. Cannabinoids and terpenes are certainly what drive a large part of the cannabis market in terms of edibles, smokable flower, vapes and extracts and the different effects and flavors that you get. We also are looking at other ingredients, like plant-based protein and hemp as a viable protein source and the ability for hemp to produce valuable fiber for textiles, as well as industrial building materials and applications.
Lastly, there are additional small molecules that we’re working on as well from a food ingredients perspective. There are all kinds of interesting compounds. Everybody talks about the cannabinoids and terpenes, but there are also things like flavonoids, and some other very interesting chemistries that we’re working on as well.
Aaron: What geographies are you currently in?
Jon: Colorado and California primarily and we have a small R&D partnership in Barcelona.

Aaron: Do you have plans for expansion beyond that?
Jon: Our current headquarters are out of Colorado, and most of our Colorado operations right now are all hemp. Our hemp business is national and international.
We work with a licensed cannabis nursery partner in California which is our primary focus for that market, but we will be expanding the cannabis genetics and nursery program into Colorado next year. From a regulated cannabis perspective, that’s the first move. Beyond that, we’re in conversations with some of the multi-state operators and cannabis brands that are emerging to talk about how to leverage our technology and our genetics platform across some of the other markets.
Aaron: How do you think about genetics in your products?
Jon: Genetics means a lot of things to different folks depending on your vantage point and where you sit in the supply chain. Our business model is based on selling plants and seeds. At the end of the day, we don’t develop oils, extracts and products specifically, but we develop the genetics behind those products.
For us, it’s not only about developing genetics that have the unique qualities or ingredients that a product company might want like CBD, or other minor cannabinoids like THCV for example, but also about making sure that those plants can be produced efficiently and effectively. The first step is to introduce the ingredient to the product. Then the second step is to make sure that growers can grow and produce the plant. That way they can stabilize their supply chain for their product line. Whether it’s for a smokable flower product, or a vape product, or an edible product, it’s really important to make sure that they can reproduce it. That’s really how we think about genetics.
Aaron: What is a smart plant? That’s something I saw on your website.
Jon: It’s really about plants that perform under specific growing regions, or growing conditions. For example, in hemp, it’s one thing to produce CBD or CBG. It’s another thing to be able to produce it efficiently in five different microclimates around the U.S. Growing hemp in Florida or Alabama down on the Gulf Coast versus growing on the Pacific Northwest coast of Washington, or Oregon are two very different growing conditions that require smart plants. Meaning they can grow and thrive in each of those conditions and still produce the intended product. Generally, the different regions don’t overlap. The genetics that you would grow in Pacific Northwest are not going to do as well as some better selected varieties for the South East.
It’s not only different outdoor growing regions, but it’s different production styles too. When you think about regulated cannabis the difference between outdoor and indoor greenhouse is mixed light production. Even with hydroponic type growing methods, there are lots of different ways to grow and produce this plant and it’s not a one size fits all. It’s really about plants that perform well, whether it’s different regions in the United States in outdoor production or different indoor greenhouses with mixed lights and production methods.
Aaron: You market CBG hemp as a product line. What made you start with CBG? Is that a pull from the market or something you guys see trending?
Jon: So I think it’s a little bit of both. We offer CBD dominant varieties and CBG dominant varieties of hemp. We also now have other cannabinoids in the pipeline that we’ll be putting out in different varieties next year. Things like CBC as well as varins, or propyl cannabinoids. Also things like CBDV, CBCV, or CBGV, which are the propylcannabinoid versions of the more familiar compounds.

There was a lot of market demand for CBG. It was a fairly easy cannabinoid to produce as a single dominant cannabinoid similar to CBD or THC. There’s a lot of up-and-coming demand for some of the other minor cannabinoids. Up until a few years ago, CBD was considered a minor cannabinoid. It wasn’t until Charlotte’s Web in the Sanjay Gupta story that it became a major cannabinoid. So I think we see some level of market pull across the category.
On the flip side of that, we have one of the world’s largest R&D teams and consolidated expertise in terms of cannabis. We see the potential for minor cannabinoids, and even terpenes and other compounds like flavonoids to have wide ranging implications in human health. Everything from wellness products, to active pharmaceutical ingredients, to recreational products. From our perspective, that’s the reason why we’re pushing these ingredients. We believe that there are a lot of good products that come out of this work and the genetics that produce these minor cannabinoids.
Aaron: Okay, great. And then last question, is there anything you’re interested in learning more about?
Jon: I think the most exciting thing for me, given my background in clinical diagnostics and human health, is to see more data around how all of these different compounds of the plant can support improved wellness, health and nutrition. I think we’ve only scratched the tip of the iceberg. This type of research and data collection takes years, even decades, especially to see outcomes over time of people using these products. I’m really excited to see more of that and also hopefully be able to make stronger conclusions about some of the benefits that can be had from this plant.
Aaron: That’s the end of the interview, thanks Jon!