As the cannabis industry grows, companies are faced with more labor related compliance and regulatory issues, which require time and expertise. Rather than hire internal staff to manage human resources (HR) and compliance, many companies choose to outsource nearly 85% of HR functions. These functions include payroll administration, HR tasks and other employment liabilities, like insurance, to a third-party Managed Service Provider (MSP). This model frees up internal resources to grow and develop the company’s core business, while also offsetting risks associated with employment, taxes and insurance.
Nicholas Murer formed WECO in 2014 to provide human capital financial services to the legal cannabis industry, offering services like payroll management, workforce management, human resource implementation, accounting solutions, recruiting and staffing. The company has since expanded to providing consulting services, financial product representation, investor asset development, wholesale trading and advising emerging market development projects worldwide.
With markets across the country maturing at a rapid rate, change is a constant. Cannabis companies operating in new markets need to maintain compliance while focusing on their business plan, which can be a difficult task. We sat down with Nick Murer to learn more about compliance issues that cannabis businesses face, like workers comp, payroll taxes, insurance and how outsourcing some HR functions can help.
Q: What are some of the major labor compliance issues faced by employers in the cannabis industry?
Nicholas Murer: Like other industries, the cannabis industry is subject to labor related regulations like paid time off, harassment prevention training, workers compensation requirements, payroll taxes, pay transparency, unemployment insurance and reporting. Unless companies have an expert, or a team of experts, monitoring and managing compliance on both the employer and employee side, they may quickly find themselves in hot water with state regulatory agencies, or even with an employee for labor law violations.
Another issue continues to be access to banking and payment processing. Many cannabis companies continue to pay employees and vendors in cash, which creates not only problems in accurate accounting, but safety concerns for employees as well. There are banks and credit unions that will work with cannabis companies, but without a partner who has relationships with and access to a proven and trusted network of banks and processors, monthly account and transaction fees can be expensive and out of reach. With the SAFE Banking Act stalled once again in Congress, this will continue to be an issue.
Q: How can cannabis companies mitigate their risk by outsourcing HR functions?
Murer: By utilizing a Managed Service Provider (MSP), cannabis companies enter a labor contract model for payroll and HR administration. An MSP is a professional workforce management company that provides comprehensive employment services for businesses.Nick Murer and the WECO team will be at the CQC this October 16-18. Click here to learn more.
This model provides the client company with the best labor practices, risk mitigation and claims management with access to national workers compensation and unemployment insurance. The MSP is the employer of record, so is responsible for most aspects of the employee/employer relationship. The day-to-day management of the employee continues with the client company, but the company’s liability and risk are reduced. MSP compliance risk reducing services include:
Background screening and reporting
Labor related compliance management at state and local levels
Handbook and policy management and distribution to employees
A central location for employee onboarding, time and assignment tracking, payroll administration and reporting
Separation of labor cost for each location/company for 280E tax mitigation
All federal, state and local tax filings
Employment verification
Employee access to health insurance
Employee access to banking for payroll direct deposit
Outsourcing to an MSP provides cost savings to the company, including:
Reduced staff
More efficient payroll processing and legal compliance
Streamlined recruitment
Utilizing WECO to manage the employer of record process allows client companies to look beyond traditional payroll to a full workforce management solution that ensures a smooth payroll and provides the tools that can support the growth and development of a workforce including compliant banking, human resources management and employee services.
About Nicholas Murer
Nicholas Murer formed WECO in 2014 to provide human capital financial services to the legal cannabis industry, offering services like payroll management, workforce management, human resource implementation, accounting solutions, recruiting and staffing. Nick Murer has more than twenty years of professional and technical sales experience working globally in the energy, engineering and scientific industries, including a substantial background in industrial technical sales, account development, marketing, human resources, acquisitions and project management. He studied accounting and business management at the University of Pittsburgh and organization development and human resource management from Colorado State University.
Vaping is a multi-billion dollar cannabis product category representing more than 20% category share in the US, according to a recent Headset.io report. The 2019 vaping crisis, whereby lung injury and several deaths were caused by the adulteration of vapor pen cartridges with vitamin E acetate, highlighted the importance of safety and emissions testing for vapor pen products. In addition to volatile organic compounds, metals and ceramics contained in the heating elements of cartridges are also a concern. While the FDA has a robust program for emissions testing in nicotine products, they do not currently regulate cannabis. Cannabis vaping is currently regulated at the state level in the United States.
Cannabis vaping is popular among minors owing to its discrete nature. In a recent study published in the Journal of the American Medical Association (JAMA), 14.7% of teens reported vaping cannabis in 2018. In a separate research study, University of Michigan researchers found that teens vaping cannabis were two times more likely to experience respiratory issues than teens who smoked e-cigarettes.
We spoke with Corey Mangold, CEO and founder of PurTec Delivery Systems, to learn more about cannabis vaping safety and their PurGuard technology. Prior to entering the cannabis space, Corey founded a software company in 1998. He also founded the advertising agency Gigasavvy in 2008, which he recently exited from in March 2021.
Aaron Green: How did you get started in the cannabis industry?
Corey Mangold: I got started in the cannabis industry in 2016. My daughter was away at college in San Luis Obispo and got pregnant and was going to have a baby, which obviously I was excited about. I decided to have her come down to Southern California and start a company together, as I’ve done multiple times in my career successfully. I wanted to show her the ropes, and teach her everything from finance to HR, to business development, marketing – everything it takes to be successful – and give her the tools that she would need to be successful for her life.
Green: What kind of things were you into before 2016?
Mangold: I founded my first company in 1998 in the software industry and had that company up until 2005. In 2008 I started another company called Gigasavvy, a nationally recognized advertising agency out of Irvine, California, which I successfully exited in March of 2021.
Corey Mangold, CEO and founder of PurTec Delivery Systems
When deciding to start a company with my daughter, we were interested in the cannabis industry – I think everybody was back in 2016. In 2016, I had started using cannabis again after probably about a 16- or 17-year hiatus. I was using a vape because I had children in the house. I went to literally anywhere I could and bought every type of cartridge on the market. What I found was that their user experience was not like what it was on the nicotine side of vaping. I reached out to associates of mine who had been manufacturing vapes since 2011, starting with the blue e-cigarette, and we engineered a unique device that was proprietary and completely unlike anything on the market. It was incredible, and still to this day, I think it’s probably the best 510 thread cart on the market. We launched that under the Orchid Essentials (CNSX: ORCD, OTC:ORVRF) brand in California and Oregon.
Green: Is that cart something that you sell to other brands as well, or is it purely for the Orchid brand?
Mangold: Yes, purely for the Orchid brand, but it’s what inspired me to start PurTec Delivery Systems. After a few years of struggling in this industry because we didn’t have the access to capital needed – Orchid is a US company traded on the Canadian Stock Exchange (CSE:ORCD, OTC:ORVRF) – and dealing in a substance that’s federally illegal, there was no access to any traditional financing, be it factoring or inventory financing. We were literally creating as much product as we could every month and then selling out almost instantly, and then waiting till the next month to get money in from all our accounts to make more. We had to slug it out. We did get into a little over 500 stores in California and Oregon, but it was just a battle, and I didn’t really want to be touching cannabis.
In 2020, I had a breakthrough in my strategy. I was watching the TV show Gold Rush and I watched one of the guys go and have to buy a new wash plant. He pulls up to this dealer’s yard that sells wash plants and tractors. I saw this dealer had a lot of inventory and clearly a lot of money, and I realized the place to make money was selling the shovels, not really digging for gold. I said to myself if I have the best shovel out there, why am I digging? I should just be innovating new shovels and selling shovels. Hence, I started PurTec Delivery Systems and now for the last year and a half have been 100% focused on developing advanced vaporizer technologies.
Green: Tell me more about PurTec.
Mangold: I founded PurTec with the sole intention of creating safe vaporizers for consumers. We conducted an 18-month safety study in Switzerland with our partners, on vaping devices in the market. I learned a lot of things that I already knew but wanted to see it proven by independent laboratories and by PhDs and MDs, and really see what was so concerning to me. For the last year and a half, we have sought to develop a safe line of vaporizers. I’m very cognizant about what’s going on in my body and want to know what’s going on internally with these products. I don’t think anyone would be using them if they knew what was really going into their lungs.
Green: What are some of the things that consumers should be thinking about when it comes to vape safety?
Mangold: Consumers should be thinking about all the different aspects from inhaling vaporized heavy metals to ceramics. Ceramic particle inhalation is one of my biggest concerns. I think it’s been ignored. I think all the manufacturers know about it and I think it’s been swept under the rug. I think it’s one of the threats that we have. There should be regulatory bodies that are out there protecting consumers like the FDA, hence why I believe federal legalization is so important, because if the FDA was involved not even one of these products would be on the market because the first thing the FDA would do would be very extensive emissions testing to find out what compounds and potential toxins are entering into your body.
Green: There’s clearly a need for safety and regulation in the space, but from where you’re sitting, is there a demand? When consumers go into a store, one of their main focuses is: what’s the THC content? How do you see consumer demand for safety and how do you think about building that awareness?
Mangold: I don’t think there is consumer demand yet. The consumer demand right now is for getting medicated and having fun or getting whatever relief or primary reason you use cannabis. I can point to a direct correlation with the opioid epidemic. No one knew they were as horrible as they are, and doctors were prescribing them left and right, and everyone thought it was okay. People think these cannabis products are okay because they’re on the shelf in every licensed dispensary, and the California Department of Health and the Department of Health in every other state and country has been involved to some degree. So, consumers think that they’re safe. The problem is they’re likely not just like we weren’t with opioids.
I don’t think the consumer demand will be there for quite some time until we start seeing a lot of long-term health impacts where we start seeing people getting lung disease, we start seeing people getting iron lung, different potential brain issues from inhaling adhesives and heavy metals. I think once the health impacts are seen clinically – just like we saw with the opioid crisis – once that was really in the forefront, everybody saw with their own eyes, and then they were aware that there was a problem. So, I think that it’s important to become aware of the potential health impacts, but I think it will take quite some time before that happens.
Green: It sounds to me like you want to get ahead of the industry on this because if it does go federally legal, there will be more stringent requirements. How do you think about that from a product design and development perspective to get ahead of a problem that exists but isn’t reflected in current regulations?
Mangold: The best thing we can do right now in the cannabis vape industry is to look at what the nicotine vape industry is doing. It is controlled by the FDA and there are standards for vaporizers in other parts of the world that are very stringent, like the AFNOR standards, which are in the European Union regulations for vaporizer safety.
What we do is we find the most stringent standards in the world, and we test our products to those standards. If the standards get stricter, we can develop our products and re-engineer them to meet those new requirements. Right now, all our products are emissions tested at AFNOR standards and over-engineered even for those standards. We also are constantly working on reduction of potentially hazardous materials: reductions of heavy metals; only using proven safe and effective materials and FDA approved materials like SAE 316L surgical stainless steel; and using improved ceramics that are not as brittle as the ceramics being used by almost every single manufacturer out there. There’s a lot of things that can be done. It takes supply chain management, understanding the technology and having strong solid teams of scientists and doctors that know this stuff much better than anyone else in the industry does, and leveraging their expertise.
Green: You recently launched a safety feature for minors. Can you tell me more about that?
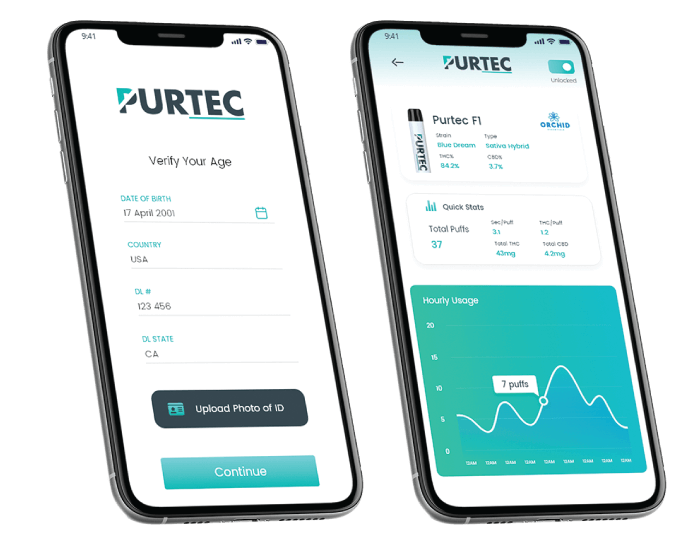
Mangold: Yes. Two weeks ago, we launched a new software application called PurGuard. PurGuard is a massive innovation and is the first of its kind that we’re aware of. It’s a piece of software that pairs with any device, whether it’s a disposable pod system or a 510 cartridge. You then pair it to your phone and take a picture of your government ID. Then the camera looks at your face, runs quick facial recognition and runs an age check through the largest age-checking platform API in the world. Then based on location and legal age of the user’s location – some states are 18 and different countries have different rules – it validates your ability in your market to be consuming that product. This technology works in 180 different countries.
Once that occurs and the device is ready for you to use, we have another feature that we’ve developed. There is an auto-lock feature that we have where if you’re a parent, like me, and you have kids in the house, you can turn your device to auto-lock right from your phone. When you walk away from your phone and are 10 feet away, your Bluetooth connection will break, and it will automatically lock the device and so your child can’t walk into your bedroom and take your device.
This technology is important to us. Consuming cannabis is horrible for the health of minors. There are serious mental effects on brain growth that occur from using cannabis at a young age because the brain is still developing up until about the age of 23 to 25. So, it’s not safe for them to be using. Of course, I’m sure we all smoked when we were in high school, but the ease of use of vape and the discretion, I think allows minors to use significantly more cannabis than previous generations did 20, 30, 40 years ago. It’s a massive problem right now and I think it’s just a matter of time before the FDA requires such protections. This industry can only survive if we protect minors. So, we’re getting ahead of the curve and setting the standard.
Green: What kind of hardware does PurGuard work with?
Mangold: PurGuard works with every single type of device that we manufacture: 510 thread cartridges, disposables, and pods. If it’s a 510-thread cartridge, the battery has to be a PurTec battery, and the cartridge has to be a PurTec cartridge. They communicate to each other through certain technologies, and it can even recognize what oils are in the cartridge or the pod or the disposable. Moreover, we can tell what strain it is, when it was manufactured, what the potency levels are and more. It records all the usage statistics. We’ve also proven with our hardware, the actual milligram contents being consumed per hit, or draw based on volume, and draw duration. We can track and report to people and say, “Hey, you’re consuming 100 milligrams of THC a day, that’s too high, you need to slow down and maybe go down to 50 milligrams a day.” That will be what is required as it is being required in the nicotine industry under the FDA pre-market tobacco applications (PMTA). When the FDA comes into cannabis, they’re going to want to see the same thing. They’re going to want to know that cannabis products are not promoting people to use more, and they are trying to get people to use less. It doesn’t mean stop using it, but use it in moderation, like everything in life. You shouldn’t be drinking a bottle of whiskey a day. You probably shouldn’t be smoking a pound of weed a day either. Everything in life is moderation and this application not only protects minors but also teaches us about our consumption habits.
Green: A theme here is “skating where the puck is going to be.” What kind of trends are you looking at right now in the industry?
Mangold: The biggest trend I see right now in the industry is disposables. We’ve seen that the trends in cannabis consumption trail behind the nicotine industry by 2-4 years. We see a lot of our customers and potential customers shifting into disposables and are now seeing a very large spike in sales of disposables. I think that’s a big trend, but with that comes another major issue: we now have lithium-ion batteries being thrown away at astonishing rates and going into landfills. PurTec has an answer for that that we’ll be launching here in the next four to six months That will be I think the biggest innovation in regards to eco-friendliness within the vape industry. That’s where I see things going right now.
Green: What are you most interested in learning about?
Mangold: The thing that interests me most, and what I’m most interested in learning about is regulations. Not the regulations themselves, but how regulations are drafted. I’ve sat in several meetings with rules committees for different regulatory bodies throughout the United States and it is laughable. I was recently in a state I’m not going to mention. I asked them what scientists and what doctors they have consulted with and they said none. I just found that dumbfounding. The state regulatory bodies are making decisions without doing due diligence and without bringing in subject matter experts in some cases.
I’m very interested in learning about how we can change our regulatory bodies. Taxpayers pay these salaries and their job at the end of the day is to protect consumers. I think that these cannabis regulatory bodies need to be way more involved with their state’s Department of Health, as well as with the FDA, and National Institute of Health and looking at this as a holistic approach. How do we protect consumers? This is a drug. It’s like anything else out there. If you’re selling tomatoes that were sprayed with a certain pesticide, you must do the research and you have to know what’s in that product before you start putting it in people’s hands. Otherwise, you may have people dying left and right. So, I’m very interested in learning more about regulatory bodies and how they need to evolve and hopefully I can help push them into evolving sooner rather than later.
Green: Great, that concludes the interview, Corey.
Hazard Analysis and Critical Control Points (HACCP) is a systematic approach that evaluates hazards that may potentially be present in food products that can harm the consumer. The process used to manufacture the product is evaluated from raw material procurement, receiving and handling, to manufacturing, distribution and consumption of the finished product1. The documented process is what is known as HACCP plan. Although HACCP was designed to evaluate hazards in foods, it can be used to assess or evaluate hazards that may potentially be present in cannabis consumable products (edibles and vaping) that can cause harm to the consumer.
HACCP plan development requires a systematic approach that covers 5 preliminary steps and 7 principles. A systematic approach means that each step must be followed as outlined. Skipping a step will result in a HACCP plan that most likely will be ineffective to control potential hazards in the product.
The 5 preliminary steps are:
Establish a HACCP team
Describe the product
Establish the intended use of the product
Develop a flow diagram
Verify the flow diagram
The 7 Principles are:
Conduct a hazard analysis
Identify the critical control points (CCPs)
Establish critical limits (CL)
Establish monitoring procedures
Establish corrective actions
Establish verification procedures
Establish records and record keeping procedures1,2
It is important to mention that HACCP plans are supported by programs and procedures that establish the minimum operational and sanitary conditions to manufacture safe products. These programs and procedures are known as pre-requisite programs (PRP) or preventative controls1,2.
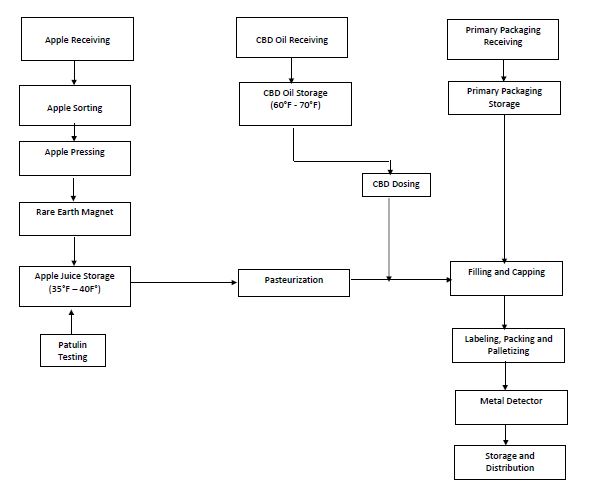
Figure 1. Flow Diagram
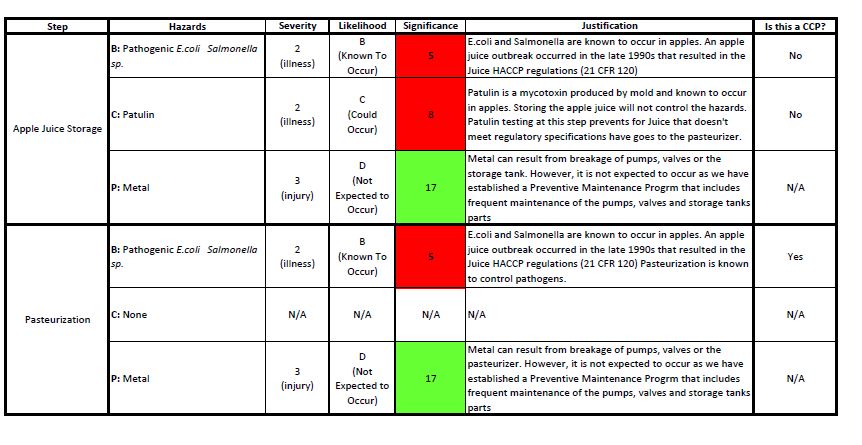
A multidisciplinary team must be established in order to ensure that all inputs of the product manufacturing process are considered during the hazards analysis discussions. The description of the product and its intended use provides detail information on ingredients, primary packaging material, methods of distribution, chemical characteristics, labeling and if any consumer might be vulnerable to the consumption of the product. A verified flow diagram is an accurate representation of the different steps followed during the product manufacturing process and will be used to conduct a hazard analysis. An inaccurate flow diagram will set the stage for an inadequate HACCP plan. Therefore, it is important that the HACCP team members verify the flow diagram. Figure 1 is a flow diagram for a fictional infused apple juice manufacturing plan that I will be using as an example.
The hazard analysis is the backbone of the HACCP plan. There are two elements that must be considered when conducting the hazard analysis:
Identification of the hazard associated with the ingredient(s) and/or the product manufacturing steps. These hazards have been categorized as: Biological, chemical (including radiological) and physical. Biological, chemical and physical hazards should be considered for each ingredient, primary packaging and process step. Also, it is important that the team is specific as to what hazard they are referring to. I often find that biological hazards are identified as “pathogens” for example. The team has to be specific on which pathogen is of concern. For example, if you are processing apple juice, the pathogens of concern are pathogenic coli and Salmonella sp. However, if you are processing carrot juice, you need to add Clostridium botulinum as a biological hazard also. If the choice of method to eliminate the hazards is pasteurization for example, the processing temperature-time combinations will differ greatly when manufacturing the apple juice vs. the carrot juice as C. botulinum is an organism that can sporulate and, therefore, is harder to kill.
Characterization of the hazard. This implies determining the significance of the potential hazard based on the severity of the consequence if it is consumed and the likelihood of occurrence in the ingredient or process step. Only steps in the process that has significant hazards should be considered further.
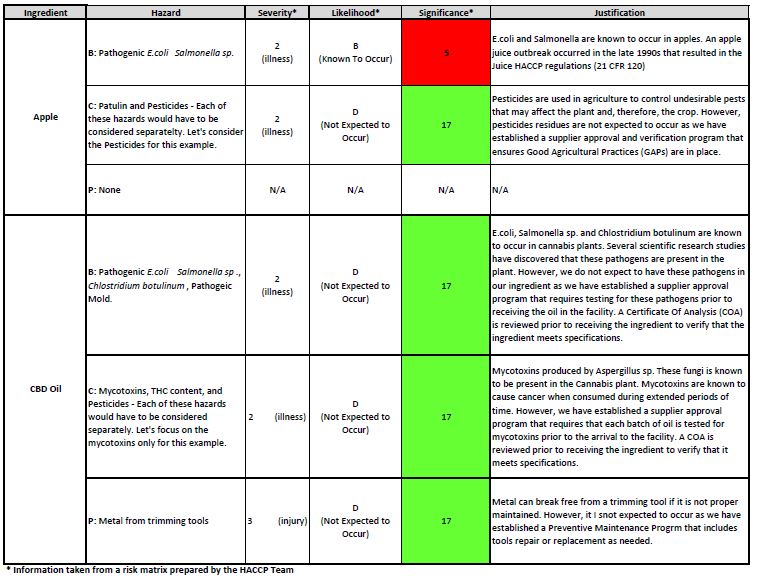
Table 1. Ingredient Hazard Analysis
In my professional experience, the hazard analysis is one of the most difficult steps to achieve because it requires the expertise of the multidisciplinary team and a lot of discussion to get to the conclusion of which hazard is significant. I find that a lot of teams get overwhelmed during this process because they consider that everything in the process may represent a hazard. So, when I am working with clients or providing training, I remind everyone that, in the bigger scheme of things, we can get stricken by a lighting in the middle of a thunderstorm. However, what will increase our chances would be whether we are close or not to a body of water for example. If I am swimming in the middle of a lake, I increase my chances to get stricken by the lighting. In comparison, if I am just sitting in my living room drinking a cup of coffee during the thunderstorm, the likelihood of being stricken by a lighting is a lot less. The same rationale should be applied when conducting the hazard analysis for manufactured products. You may have a hazard that will cause illness or death (high on the severity chart) but you also may have a program that mitigates the likelihood of introducing or having the hazard. The program will reduce the significance of the hazard to a level that may not need a critical control point to minimize or eliminate it.
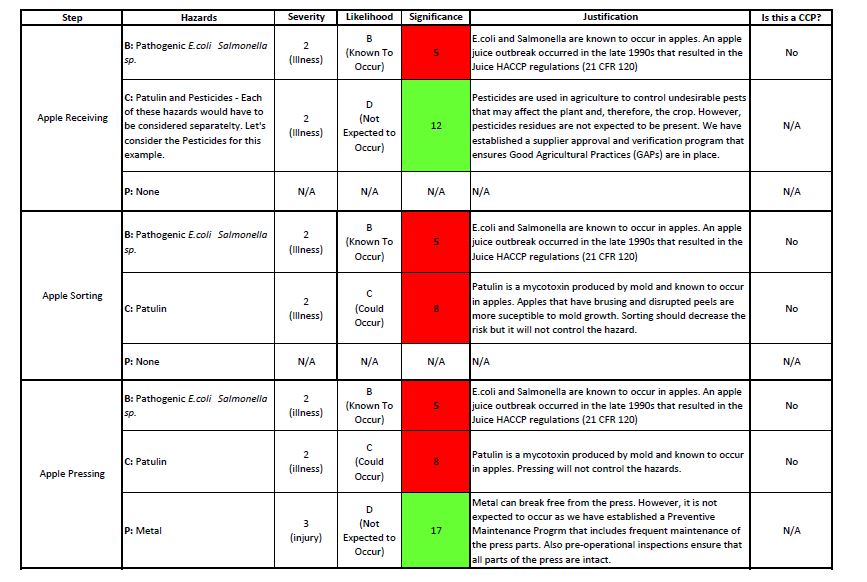
Table 2. Process Hazard Analysis (1)
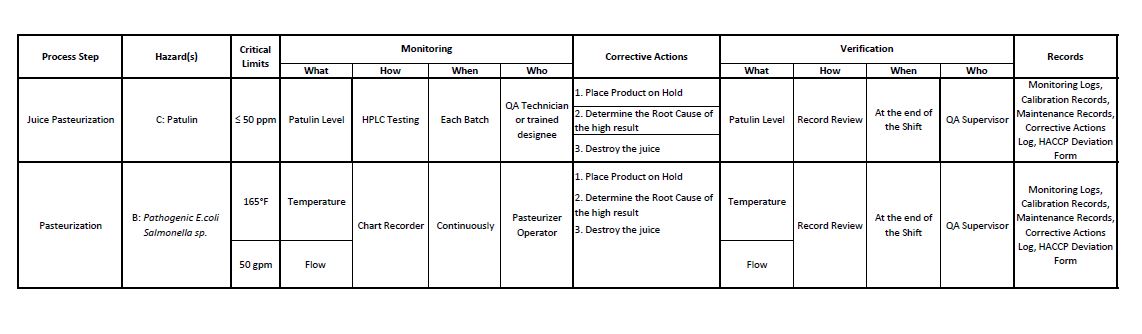
Clear as mud, right? So, how would this look like on the infused apple juice example? Table 1 shows the hazard analysis for the ingredients. Tables 2 and 3 show the hazard analysis for the part of the process. In addition, I have identified the CCPs: Patulin testing and pasteurization. There is a tool called the CCP decision tree that is often used to determine the CCPs in the process.
Once we have the CCPs, we need to establish the critical limits to ensure that the hazard is controlled. These limits must be validated. In the case of Patulin, the FDA has done several studies and has established 50 ppm as the maximum limit. In the case of pasteurization, a validation study can be conducted in the juice by a 3rd party laboratory. These studies typically are called thermal death studies (TDS) and provide the temperature and time combination to achieve the reduction of the pathogen(s) of concern to an acceptable level that they do not cause harm. In juice, the regulatory requirement is a 5-log reduction. So, let’s say that the TDS conducted in the infused apple juice determined that 165°F for 5 seconds is the critical limit for pasteurization. Note that the 5 seconds will be provided by the flow of the product through the holding tube of the pasteurizer. This is measured based on flow in gallons per minute.
Table 3. Process Hazard Analysis (2)
Monitoring is essential to ensure that the critical limits are met. A monitoring plan that outlines what, how, when and who is responsible for the monitoring is required.
Ideally, the system should not fail. However, in a manufacturing environment, failures can happen. Therefore, it is important to pre-establish steps that will be taken to ensure that the product is not out of the control of the facility in the event of a deviation from the HACCP plan. These steps are called corrective actions and must be verified once they are completed. Corrective actions procedures must address the control of the product, investigation of the event, corrective actions taken so the deviation doesn’t reoccur and product disposition.
Table 4. HACCP Plan Summary
Verification activities ensure that the HACCP plan is being followed as written. Typically, verification is done by reviewing the records associated with the plan. These records include but are not limited to monitoring records, calibration records, corrective action records, and preventive maintenance records for equipment associated with the CCPs. Record review must be done within 7 working days of the record being produced.
Finally, establishing records and record keeping procedures is the last step on developing HACCP plans. Records must be kept in a dry and secure location.
Table 4 show the summary of the HACCP plan for the infused apple juice example.
For more information on how to develop a HACCP plan for your facility, read the resources below:
According to a press release emailed today, Perry Johnson Laboratory Accreditation Inc. (PJLA) announced the accreditation of PharmLabs LLC to ISO/IEC 17025. Based in San Diego, California, PharmLabs has four locations, with three in the Southern California region and one in Maui.
PharmLabs offers a very wide variety of services including: California Compliance testing, a specialized Hemp Testing Program, Hemp Biomass Verification testing, and THC-free testing for the state of California. In addition, they offer the testing of cannabinoids, pesticides, residual solvents, microbiological contaminants, mycotoxins, heavy metals, terpenes, water activity, moisture content, and filth/foreign material testing.
“Our experience with [Perry Johnson Laboratory Accreditation] has been incredible over the years. Since we have multiple locations, we have had many visits with PJLA and their knowledge and quick response time has helped us get where we need to be,” says Greg Magdoff, founder of PharmLabs.
According to the press release, PharmLabs has plans to expand throughout the state of California and the rest of the United States in both hemp and cannabis testing in legal states.
The CannTrust story may have shocked the uninitiated, but it hit almost every bogeyman the legitimizing industry has both feared and suffered from, particularly of late.
Here, generally, is the issue. Especially in Europe (even more especially in places like Germany, the UK and other emerging markets), budding cannapreneurs need each other. A distributor in Germany, for example, cannot get their final (federal) licenses allowing them to do business without establishing a relationship with an existing producer. That producer also needs relationships with established distributors to get their licenses.
In a fraught world, where all parties are evolving rapidly (and this also includes the “Big Boys” from Canada and several U.S. states including California), supply chain logistics, and even contract agreements if not licensing beyond that requires a level of honesty, integrity and transparency the industry, largely has not achieved yet.
That said, there are also parties, if not individuals and companies determined to set themselves on the straight and narrow – and play by the emerging “rules” – and then there are also clearly companies which, well, do not.
Being out of compliance, at any step of the chain, including when your product is sold via government agencies, is already a recipe for disaster.What this brave new world of cannabis requires, however, and from everyone – from grower, to manufacturer, packager, distributor and service delivery – is that all ecosystem partners must be in compliance.
Ensuring that can be a full time job. But what it also means is that to have a fully compliant product, every party in the chain bears responsibility for upholding standards that so far have proved hard to reach for many.
The time has come, in other words, where that is no longer an option.
The First Step Is Certification…
In a world where every member of the diverse cannabis ecosystem requires certification, determining what, and from whom is the first hurdle – both for buyer and seller. If one has GMP-certified product, that is awesome. But there are also treaties in the room that only allow some GMP certifications to be considered equal to others. If you are in Lesotho right now, for example, far from Europe, your biggest concern is not just looking to the EU but figuring out a way to export your crop into your neighbouring (and surrounding) country – namely South Africa.
This example, while seemingly far away, in fact, is the biggest bugbear in determining who can sell to whom even within Europe (let alone countries just outside and far beyond the region).
Determining cert presence, if not validity, however, is only the tip of the iceberg. And depending on who you are, that path alone is not a one time dalliance with authorities, but multiple certifications that must all also be kept current.
But It is Not The Only One…
The second hurdle, of course, is also checking the verity of everyone you do business with. For a producer, this includes making sure that processing, packaging, and even transportation are in compliance. In Canada, of course, this has been short circuited by the ability of producers to ship directly to patients.
In Europe, however, this is far from the case. And that is also why the entire conversation is also getting not only much more granular, but expensive. Pharmaceutical regulations are actually what guide the rules of the road here.
Walking floors, and checking, in person, may or not be mandated by international treaties at this point. However, most of the young producers on the ground here are implementing policies of personal visits to their vendors. In Massachusetts of late, this is also on the drawing board. Albeit on a “state” level, the reality is that both federal, state and more local training is a watchword, if not a must, now on the roadmap.
Being out of compliance, at any step of the chain, including when your product is sold via government agencies, is already a recipe for disaster.
And while that obviously is a challenge, companies must step up to the plate internally to commit to the same. It is too dangerous to ignore such steps. Including the easy to reach ones, like staff background checks and decent cybersecurity safeguards. The former has blown several enterprising cannadudes out of the driver’s seat already in Europe over the last few years. The latter is an emerging threat in a region that is also home to GDPR regulation (and growing fines).
For that very reason, certainly on the ground in Germany if not across Europe and in those countries and companies that wish to supply the same, supply chain verification, that is constant, consistent and verifiable, is the path for the industry both as of now and in the immediate future.
Jason Neely founded Stratos in 2014, when he and a small group of people left the pharmaceutical industry in search of a new endeavor in the cannabis marketplace. The concept was straightforward: Apply pharmaceutical methodologyof production to cannabis products. Back then, Stratos offered a range of THC-infused tabletsin the Colorado market.
Brenda Verghese, vice president of research & development
Brenda Verghese, vice president of research & development, was one of five people on staff when Stratos launched. Now they have about 30 team members. Consumers were looking for a cannabis product that would be consistent and reliable every time, taking the guesswork out of infused products dosage. That’s where Brenda Verghese found her skillset useful.
Transitioning to the pharmaceutical industry right out of college, Verghese started her career as a chemist and worked her way up to the R&D business development sector. “I specializedin formulations and taking a product from concept to commercialization in the pharmaceutical space,” says Verghese. “Jason Neely approached me with the idea of a cannabis company and focusing on making products as effective and consistent as possible, so really bringing pharmaceutical science into the cannabis space. In the matter of 4 years we grew substantially, mainly focusing on the efficacy of products.”
Behind the scenes at packaging and labeling Image credit: Lucy Beaugard
Soon after the success of their THC products became apparent, Stratos launched a CBD line, quickly growing their portfolio to include things like tinctures and topicals as well. According to Verghese, they are hoping that what’s been established on the THC side of their business as far as reproducibility and consistency is something that consumers will also experience on the CBD side. “Quality and consistency have definitely driven our growth,” says Verghese. “That is what consumers appreciate most- the fact that every tablet, tincture or swipe of a topical product is going to be consistent and the same dose every time.” This is what speaks to their background in the pharmaceutical sciences, FDA regulation has taught the Stratos team to create really robust and consistent formulations.
Quality in manufacturing starts at the source for Stratos: their suppliers. They take a hard look at their supply of raw materials and active ingredients, making sure it meets their standards. “The supplier needs to allow us to do an initial audit and periodic audits,” says Verghese. “We require documentation to verify the purity and quality of oil. We also do internal testing upon receipt of the materials, verifying that the COAs [certificates of analysis] match their claims.”
Process validation in action at the Stratos facility (image credit: Lucy Beaugard)
Verghese says maintaining that attention to detail as their company grows is crucial. They implement robust SOPs and in-process quality checks in addition to process testing. They test their products 5-6 times within one production batch. Much of that is thanks to Amy Davison, director of operations and compliance, and her 15 years of experience in quality and regulatory compliance in the pharmaceutical industry.
Product testing alone cannot assess quality for an entire lot or batch of product; therefore, each step of the manufacturing process must be controlled through Good Manufacturing Practices (GMP). Process validation is an aspect of GMPs used by the pharmaceutical industry to create consistency in a product’s quality, safety and efficacy. There are three main stages to process validation: process design, process qualification and continued process verification. Implementing these stages ensures that quality, including dosing accuracy, is maintained for each manufactured batch of product.
Fast forward to today and Stratos is looking at expanding their CBD products line significantly. While their THC-infused products might have a stronger brand presence in Colorado, the CBD line offers substantial growth potential, given their ability to ship nationwide as well as online ordering. “We are always evaluating different markets and looking for what suits Stratos and our consumer base,”says Verghese.
By Ravi Kanipayor, Christian Bax, Dr. George Anastasopoulos No Comments
As state cannabis regulatory frameworks across the country continue to evolve, accreditation is becoming increasingly important. Because it provides consistent, turnkey standards and third-party verification, accreditation is quickly emerging as an important tool for regulators. For cannabis testing laboratories, this trend has been especially pronounced with the increasing number of states that require accreditation to ISO/IEC 17025.
As of 2017 there were nearly 68,000 laboratories accredited to ISO/IEC 17025, making it the single most important benchmark for testing laboratories around the world. ISO/IEC 17025:2005 specifies the general requirements for the competence to carry out tests including sampling. It covers testing performed using standard methods, non-standard methods and laboratory-developed methods. It is applicable to all organizations performing tests including cannabis labs. The standard is applicable to all labs regardless of the number of personnel or the extent of the scope of testing activities. Developed to promote confidence in the operation of laboratories, the standard is now being used as a key prerequisite to operate as a cannabis lab in many states.
There are currently 26 states in the United States (also Canada) that require medical or adult-use cannabis to be tested as of February 2019. Of those states, 18 require cannabis testing laboratories to be accredited – with the vast majority requiring ISO/IEC 17025 accreditation. States that require testing laboratories to attain ISO/IEC 17025 accreditation represent some of the largest and most sophisticated cannabis regulatory structures in the country, including California, Colorado, Maryland, Massachusetts, Michigan, Nevada and Ohio. As a consequence, many cannabis testing laboratories are taking note of recent changes to ISO/IEC 17025 standards.
ISO/IEC 17025 was first issued in 1999 by the International Organization for Standardization. The standard was updated in 2005, and again in 2017. The most recent update keeps many of the legacy standards from 2005, but adds several components – specifically requirements for impartiality, risk assessment and assessing measurement uncertainty. The remainder of this article takes a deeper dive into these three areas of ISO/IEC 17025, and what that means for cannabis testing laboratories.Objectivity is the absence or resolution of conflicts of interest to prevent adverse influence on laboratory activities.
Impartiality
ISO/IEC 17025:2005 touched on an impartiality requirement, but only briefly. The previous standard required laboratories that belonged to organizations performing activities other than testing and/or calibration to identify potential conflicts of interest for personnel involved with testing or calibration. It further required that laboratories had policies and procedures to avoid impartiality, though that requirement was quite vague.
ISO/IEC17025:2017 emphasizes the importance of impartiality and establishes strict requirements. Under the new standard, labs are responsible for conducting laboratory activities impartially and must structure and manage all laboratory activities to prevent commercial, financial or other operational pressures from undermining impartiality. The definitions section of the standard defines impartiality as the “presence of objectivity.” Objectivity is the absence or resolution of conflicts of interest to prevent adverse influence on laboratory activities. For further elaboration, the standard provides similar terms that also convey the meaning of impartiality: lack of prejudice, neutrality, balance, fairness, open-mindedness, even-handedness, detachment, freedom from conflicts of interest and freedom from bias.
To comply with the new standard, all personnel that could influence laboratory activities must act impartially. ISO/IEC 17025:2017 also requires that laboratory management demonstrate a commitment to impartiality. However, the standard is silent on how labs must demonstrate such commitment. As a starting point, some cannabis laboratories have incorporated statements emphasizing impartiality into their employee handbooks and requiring management and employee training on identifying and avoiding conflicts of interest.
Risk Assessment
Both the 2005 and 2017 versions contain management system requirements. A major update to this is the requirement in ISO/IEC 17025:2017 that laboratory management systems incorporate actions to address risks and opportunities. The new risk-based thinking in the 2017 version reduces prescriptive requirements and incorporates performance-based requirements.
Under ISO/IEC 17025:2017, laboratories must consider risks and opportunities associated with conducting laboratory activities. This analysis includes measures that ensure that:
The lab’s management system is successful;
The lab has policies to increase opportunities to achieve its goals and purpose;
The lab has taken steps to prevent or reduce undesired consequences and potential failures; and
The lab is achieving overall improvement.
Labs must be able to demonstrate how they prevent or mitigate any risks to impartiality that they identify.To comply with ISO/IEC 17025:2017, labs must plan and implement actions to address identified risks and opportunities into management systems. They must also measure the effectiveness of such actions. Importantly, the standard requires that the extent of risk assessments must be proportional to the impact a given risk may have on the validity of the laboratory’s test results.
ISO/IEC 17025:2017 does not require that labs document a formal risk management process, though labs have discretion to develop more extensive methods and processes if desired. To meet the requirements of the standard, actions to address risks can include sharing the risk, retaining the risk by informed decision, eliminating the risk source, pinpointing and avoiding threats, taking risks in order to pursue an opportunity, and changing the likelihood or consequence of the risk.
ISO/IEC 17025:2017 references “risks” generally throughout most of the standard. However, it specifically addresses risks to a laboratory’s impartiality in section 4.1. Note, the new standard requires that labs must not only conduct activities impartially, but also actively identify risks to their impartiality. This requirement is on-going, not annually or bi-annually. Risks to impartiality include risks arising from laboratory activities, from laboratory relationships, or from relationships of laboratory personnel. Relationships based on ownership, governance, shared resources, contracts, finances, marketing, management, personnel and payment of a sales commission or other inducements to perform under pressure can threaten a laboratory’s impartiality. Labs must be able to demonstrate how they prevent or mitigate any risks to impartiality that they identify.
Assessing Measurement Uncertainty With Decision Rules
ISO/IEC 17025:2005 required (only where necessary and relevant) test result reports to include a statement of compliance/non-compliance with specifications and to identify which clauses of the specification were met or not met. Such statements were required to take into account measurement uncertainty and if measurement results and uncertainties were omitted from the statement, the lab was required to record and maintain the results for future reference.
ISO/IEC 17025:2017 requires similar statements of conformity with an added “decision rule” element. When statements of conformity to a specification or standard are provided, labs must record the decision rule it uses and consider the level of risk the decision rule will have on recording false positive or negative test results. Like the 2005 version, labs must include statements of conformity in test result reports (only if necessary and relevant- see 5.10.3.1 (b)). Now, test result reports on statements of conformity must include the decision rule that was employed.
Moving Forward
Because many states require ISO/IEC 17025 accreditation for licensing, cannabis testing labs across the country would be well advised to closely monitor the implications of changes in ISO/IEC 17025:2017 related to impartiality, risk assessment and measurement uncertainty. If you run a cannabis testing lab, the best way to ensure compliance is education, and the best place to learn more about the new requirements is from a globally recognized accreditation body, especially if it is a signatory to the International Laboratory Accreditation Cooperation (ILAC) for testing laboratories, calibration laboratories and inspection agencies.
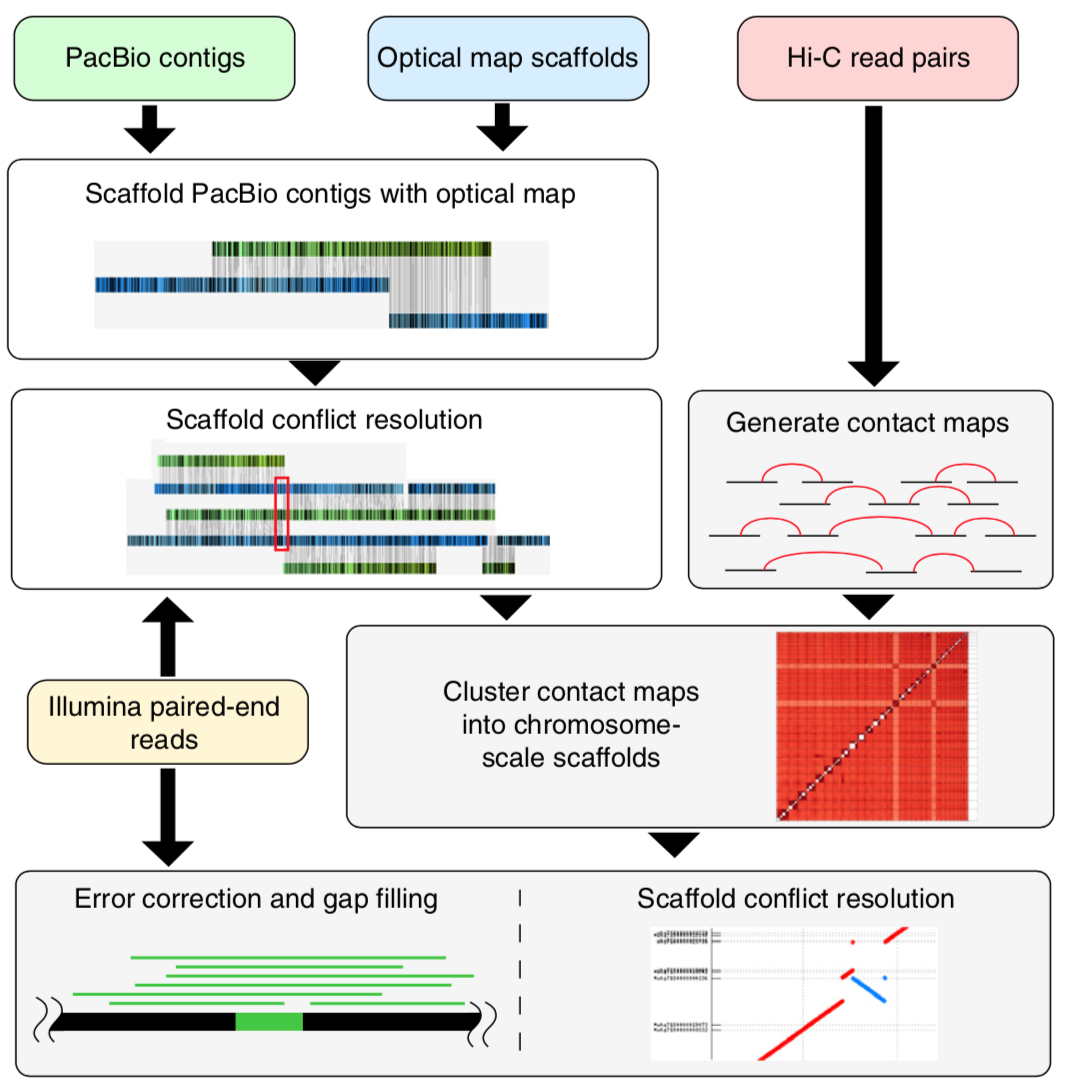
Genome sequencing has made remarkable strides since the initiation of “The Human Genome Project” in 1990. Still, there are many challenges that must be overcome before this methodology can reach its fullest potential and be useful in serving as a method of Cannabis sativa genetics verification and tracking throughout the cannabis supply chain. Several major milestones that must be realized include end-to-end haploid type (single, unpaired set of chromosomes instead of complete paired set or “diploid”), long read, resolved genome sequences at a reasonable cost within a reasonable timeframe and with confidence in accuracy (Mostovoy et al.). These genomes are typically generated as shorter reads that are then scaffolded (Fig 1.) or matched to reference genomes in order to build a longer continuous read. While shorter sequencing reads indeed lower the cost barrier for producing more genomic data, it has created another issue as a result of this short-read technology.
Figure 1: Four sets of sequencing data (long-read WGS, Hi-C, optical mapping, and short-read WGS) were produced to generate the goat reference genome. A tiered scaffolding approach using optical mapping data followed by Hi-C proximity-guided assembly produced the highest-quality genome assembly. (Bickhart et al.)
There are two main issues with the more affordable short read sequencing methodology, the first being that sequential variants are typically not detected, especially if they involve a ton of repeats/inverted repeats, due to the limitation of the current referenced Cannabis genomes and the mapping process of the short-read sequences. This is especially unfortunate because larger variants can have up to a 13% variance within a diploid multichromosomal genome, such as Cannabis sativa, and this variance is thought to largely contribute to disease in various species, or maybe terpene profile in Cannabis sativa. Not being able to detect these variances with more affordable sequencing methodologies is particularly problematic and reference genomes produced with short read sequences are typically highly fragmented. The second limitation is the inherent errors, gaps and other ambiguities associated with taking tons of short read sequences and combining them all, like a jigsaw puzzle, in order to draft the larger genomic picture. While there is software with algorithms to assist in deciphering raw sequences, there is still much more work to be done on this challenge, considering that cannabis genome sequencing is new genomics territory. Unfortunately, as researchers seek higher and higher levels of data quality, shortcomings of this type of sequencing technology begin to become apparent. This sort of sequencing methodology relies heavily on reference sequences. This isn’t much of an issue with microbial genomes, which tend to be rather short and typically have one chromosome, however, when seeking to analyze much longer genomes with multiple diploid chromosomes and tons of mono and dinucleotide repeats, problems arise (English et al.).
Figure 2: Blockchain Digital Stamping Certificate which publicly documents the date and time of the completion of this work. (Mckernan – Crypto Funded Public Genomics)
The other category of sequencing is long read sequencing. Long read sequencing is as it sounds, the deciphering of much longer DNA strands. Of course, the technology is limited by the quality of the DNA captured, therefore, special high molecular weight DNA extraction protocols must be deployed in order to obtain the proper DNA quality (Fig. 3). Once this initial limitation is overcome there is the stark cost of long read sequencing technology. PacBio without a doubt makes one of the highest quality long read sequence generating instruments that has ever graced the field of biotechnology, but due to the steep price tag of the machine, progress in this field has been stifled simply because it just isn’t affordable and the read depth for mammalian and plant genomes is currently almost completely prohibitive until read lengths double in length for this instrumentation. In order to produce what is considered to be a “validated genome” both short read and long read sequencing methodologies are combined. Long read sequencing data is used to produce the reference contigs because they are much easier to assemble, then short read sequencing is scaffolded against the reference contigs as a sort of “consensus validation” of the long read contigs.
Figure 3: Depiction of various DNA high molecular weight DNA quality captured during cannabis genome submission project. (Mckernan – Crypto Funded Public Genomics)
Despite the shortcoming of utilizing short read sequencing technology for analysis of the cannabis genome, it is still useful especially when combined with other longer read sequencing technologies or optical mapping technologies. Kevin McKernan, chief scientific officer of Medicinal Genomics, has been working feverishly to bridge the information gap between the cannabis genome and other widely studied plant genomes. As a scientist that worked on the Human Genome Project in 2001, McKernan has a demonstrated history of brilliance in the field of genomics. This paved the way for him to coordinate the first crypto funded and blockchain notarized sequencing project (DASH DAO funded) (Fig. 2), which was completed in 60 days, and surprisingly showed that the cannabis genome is over 1 billion bases long which is 30% larger than any cannabis genome submitted prior to his work. By reaching the standard of 500kb N50 set forth by the Human Genome Project, Kevin McKernan was able to see new aspects of the cannabis genome that were not visible due to the fragmented genomic data previously generated. Information such as a possible linkage of THCA synthase and CBDA synthase genes is crucial when seeking to use the cannabis genome for verification and tracking purposes. This is because special linkages can be considered a type of “genetic marker” that may be used to differentiate cannabis cultivars and lineages. There are many types of genetic markers, including SNP (single nucleotide polymorphisms), VNTR (variable number tandem repeats) and even patterns of gene expression. Funding and recording of cannabis genomics must be further developed in order for potential markers to be identified and validated via larger scale genome-wide association studies.
These technologies, when combined, often reduce the number of scaffolds while increasing the percent of resolved genome by filling in gaps within the drafted genome. Nanopore sequencing is an especially interesting and innovative sequencing technology that is useful in many ways. One of the most powerful uses of this technology is its ability to upgrade the quality of draft and pushed genomes by resolving poorly organized genomes and genomic structure for a fraction of the time and cost of other long read sequencing platforms (Jian et al.), making it an excellent candidate for solving cost and time constraints. Nanopore’s portability and convenience makes it a real-time solution to solving genetics-based problems and questions. A notable use of this technology is recorded during an epidemiological outbreak in Africa, its proof of concept in pathogen detection in space, and its ability to detect base modifications during sequencing process. Even still there are more uses to this exciting technology and it has the potential to elevate cannabis genomics and the field of genomics entirely, while remaining portable and expeditious. A shortcoming of the Nanopore sequencing platform is its low sequencing coverage, which makes this platform inefficient for applications like haplotype phasing and single nucleotide variant detection due to the number of variants to be detected being smaller than the published variant-detection error rates of algorithms using MinION data. Single nucleotide variants can be considered to be genetic markers, especially markers for disease, so this is what inhibits Nanopore from resolving our cannabis genome sequencing problems, as of today.
There are genetic markers to discover, molecular biology protocols to optimize, and industry wide potential for exciting collaborationMany algorithmic problems seem to occur due to input data quality. Typical input data quality suffers as the reads get longer and the sequencing depth gets shorter, resulting in not enough data being generated by the sequencing to provide confidence in the genome assembly. To mitigate this, scientists may decide to fractionate a genome, sequence it, or they may clone a difficult to sequence region with highly repetitive regions in order to produce reads with greater depth and thus resolve the region. They can then perform single molecule sequencing to resolve genome structure then determine and confirm the place of the cloned region. Thus, it seems that the best solution to the limitation of algorithms is to be aware of sequencing platform limitations and compensate for these limitations by using more than one sequencing platform to obtain enough pertinent data to confidently produce authentic, “validated” genome assemblies (Huddleston et al.). With input data being critical in producing accurate sequencing data, standardization of DNA isolation protocols, extraction reagents and any enzymes utilized may be deemed necessary.
To conclude, the field of cannabis genomics is teeming with opportunities. There are genetic markers to discover, molecular biology protocols to optimize, and industry wide potential for exciting collaboration. More states will need to take into account the lack of federal government research grant availability and begin to think of creative ways to get cannabis science funds to continue the development of this industry. Specifically speaking, developing a feasible method for genetic tracking of cannabis plants will require improvements within the availability of sequencing technology, improvements in deploying the resources to these projects in order for them to be completed expeditiously, and standardization/validation of methods and SOPs used in order to increase confidence in the accuracy of the data generated.
A special thank you to all of my cannabis industry mentors that have molded and elevated my understanding of current needs and applied technologies within the cannabis industry, without you there would be no career within this industry for me. You are immensely appreciated.
Citations
Bickhart, D. M., Rosen, B. D., Koren, S., Sayre, B. L., Hastie, A. R., Chan, S., . . . Smith, T. P. (2017). Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nature Genetics,49(4), 643-650. doi:10.1038/ng.3802
English, A. C., Salerno, W. J., Hampton, O. A., Gonzaga-Jauregui, C., Ambreth, S., Ritter, D. I., . . . Gibbs, R. A. (2015). Assessing structural variation in a personal genome—towards a human reference diploid genome. BMC Genomics,16(1). doi:10.1186/s12864-015-1479-3
Huddleston, J., Ranade, S., Malig, M., Antonacci, F., Chaisson, M., Hon, L., . . . Eichler, E. E. (2014). Reconstructing complex regions of genomes using long-read sequencing technology. Genome Research,24(4), 688-696. doi:10.1101/gr.168450.113
Jain, M., Olsen, H. E., Paten, B., & Akeson, M. (2016). The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biology,17(1). doi:10.1186/s13059-016-1103-0
Mostovoy, Y., Levy-Sakin, M., Lam, J., Lam, E. T., Hastie, A. R., Marks, P., . . . Kwok, P. (2016). A hybrid approach for de novo human genome sequence assembly and phasing. Nature Methods,13(7), 587-590. doi:10.1038/nmeth.3865
In Part 3 of this series on HACCP, Critical Control Points (CCPs), validation of CCPs and monitoring of CCPs were defined. When a HACCP plan identifies the correct CCP, validates the CCP as controlling the hazard and monitors the CCP, a potential hazard is controlled in the manufacturing and packaging of cannabis-infused edibles. The food industry is big on documentation. If it’s not documented, it did not happen. The written hazard analysis, validation study and monitoring of CCPs create necessary records. It is these records that will prove to a customer, auditor or inspector that the edible is safe. Here in Part 4, more recordkeeping is added on for deviation from a CCP, verification and a recall plan.
Take Corrective Action When There Is a Deviation from a Critical Control Point
Your food safety team conducts a hazard analysis, identifies CCPs and decides on monitoring devices, frequency and who is responsible for monitoring. You create an electronic or paper record of the monitoring for every batch of edible to document critical limits were met. Despite all your good efforts, something goes wrong. Maybe you lose power. Maybe the equipment jams. Nothing is perfect when dealing with ingredients, equipment and personnel. Poop happens. Because you are prepared before the deviation, your employees know what to do. With proper training, the line worker knows what to do with the equipment, the in-process product and who to inform. In most cases the product is put on hold for evaluation, and the equipment is fixed to keep running. The choices for the product include release, rework or destroy. Every action taken needs to be recorded on a corrective action form and documents attached to demonstrate the fate of the product on hold. All the product from the batch must be accounted for through documentation. If the batch size is 100 lb, then the fate of 100 lb must be documented.
Verify Critical Control Points Are Monitored and Effective
First, verification and validation are frequently confused by the best of food safety managers. Validation was discussed as part of determining CCPs in Part 3. Validation proves that following a CCP is the right method for safety. I call validation, “one and done.” Validation is done once for a CCP; while verification is ongoing at a CCP. For example, the time and temperature for effective milk pasteurization is very well known and dairies refer to the FDA Pasteurized Milk Ordinance. Dairies do not have to prove over and over that a combination of time and temperature is effective (validation), because that has been proven.
I encourage you to do as much as you can to prepare for a recall.A CCP is monitored to prove the safety parameters are met. Pasteurization is an example of the most commonly monitored parameters of time and temperature. At a kill step like pasteurization, the employee at that station is responsible for accurate monitoring of time and temperature. The company managers and owners should feel confident that CCPs have been identified and data are being recorded to prove safety. Verification is not done by the employee at the station but by a supervisor or manager. The employee at the station is probably not a member of the food safety team that wrote the HACCP plan, but the supervisor or manager that performs verification may be. Verification is proving that what was decided by the food safety team is actually implemented and consistently done.
Verification is abundant and can be very simple. First, every record associated with a CCP is reviewed by a supervisor or manager, i.e. someone who did not create the record. This can be a simple initial and date at the bottom of the record. Every corrective action form with its associated evaluation is verified in the same way. When HACCP plans are reviewed, that is verification. Verification activities include 1) testing the concentration of a sanitizer, 2) reviewing Certificates of Analysis from suppliers, 3) a review of the packaging label and 4) all chemical and microbiological testing of ingredients and product. The HACCP plan identifies CCPs. Verification confirms that implementation is running according to the plan.
Verification is like a parent who tells their child to clean their room. The child walks to their room and later emerges to state that the room is clean. The parent can believe the word of the child, if the child has been properly trained and has a history of successfully cleaning their room. At some frequency determined by the parent, the room will get a parental visual check. This is verification. In the food industry, CCP monitoring records and corrective action must be reviewed within seven days after the record is created and preferably before the food leaves the facility. Other verification activities are done in a timely manner as determined by the company.
Product recalls due to manufacturing errors in sanitation cause mistrust among consumers.
Write a Recall Plan
In the food industry, auditors and FDA inspectors require a written recall plan. Mock recalls are recommended and always provide learning and improvement to systems. Imagine your edible product contains sugar, and your sugar supplier notifies you that the sugar is recalled due to glass pieces. Since you are starting with the supplier, that is one step back. Your documentation of ingredients includes lot numbers, dates and quantity of sugar.You keep good records and they show you exactly how much of the recalled lot was received. Next you gather your batch records. Batches with the recalled sugar are identified, and the total amount of recalled sugar is reconciled. You label every batch of your edible with a lot code, and you identify the amount of each affected lot and the customer. You have a press release template in which you add the specific information about the recall and affected lots. You notify every customer where the affected edible was shipped with a plan to return or destroy the edible. When you notify your customers, you go one step forward.
How would your company do in this situation? I have witnessed the difficulties a company faces in a recall when I was brought in to investigate the source of a pathogen. Food safety people in my workshops who have worked through a recall tell me that it was the worst time of their life. I encourage you to do as much as you can to prepare for a recall. Here are two good resources:
Parts One and Two in this series have defined Good Manufacturing Practices, introduced Hazard Analysis and Critical Control Points (HACCP) and explained the first HACCP step of hazard analysis. A food safety team will typically work from a flow diagram to identify biological, chemical or physical hazards at each step of processing and packaging. Once the hazard is identified, the severity and probability are debated. Hazards with severe consequences or high probability are carried through the HACCP plan as Critical Control Points (CCPs).
Critical Control Points definedHACCP is a do-it-yourself project.
Where exactly will the hazard be controlled? CCPs are embedded within certain steps in processing and packaging where the parameters, like temperature, must be met to ensure food safety. Failure at a CCP is called a deviation from the HACCP plan. The food safety team identifies where manufacturing problems could occur that would result in a product that could cause illness or injury. Not every step is a CCP! For example, I worked with a client that had several locations for filters of a liquid stream. The filters removed food particles, suspended particulates and potentially metal. We went through a virtual exercise of removing each filter one-by-one and talking through the result on controlling the potential hazard of metal. We agreed that failure of the final filter was the CCP for catching metal, but not the other filters. It was not necessary to label each filter as a CCP, because every CCP requires monitoring and verification.
Identification of a CCP starts more documentation, documentation, documentation.
Do you wish you had more reports to write, more forms to fill out, more data to review? No. Nobody wants more work. When a CCP is identified, there is more work to do. This just makes sense. If a CCP is controlling a hazard, you want to know that the control is working. Before I launch into monitoring, I digress to validation.
CCP validationThis is where someone says, “We have always done it this way, and we have never had a problem.”
You want to know if a critical step will actually control a hazard. Will the mesh of a filter trap metal? Will the baking temperature kill pathogens? Will the level of acid stop the growth of pathogens? The US had a major peanut butter recall by Peanut Corporation of America. There were 714 Salmonella cases (individuals) across 46 states from consumption of the contaminated peanut butter. Imagine raw peanuts going into a roaster, coming out as roasted peanuts and being ground into butter. Despite the quality parameters of the peanut butter being acceptable for color and flavor, the roasting process was not validated, and Salmonella survived. Baking of pies, pasteurization of juice and canning all rely on validated cook processes for time and temperature. Validation is the scientific, technical information proving the CCP will control the hazard. Without validation, your final product may be hazardous, just like the peanut butter. This is where someone says, “We have always done it this way, and we have never had a problem.” Maybe, but you still must prove safety with validation.
The hazard analysis drives your decisions.
Starting with the identification of a hazard that requires a CCP, a company will focus on the control of the hazard. A CCP may have one or more than one parameter for control. Parameters include time, temperature, belt speed, air flow, bed depth, product flow, concentration and pH. That was not an exhaustive list, and your company may have other critical parameters. HACCP is a do-it-yourself project. Every facility is unique to its employees, equipment, ingredients and final product. The food safety team must digest all the variables related to food safety and write a HACCP plan that will control all the hazards and make a safe product.
Meeting critical limits at CCPs ensures food safety
The HACCP plan details the parameters and values required for food safety at each CCP.The HACCP plan identifies the minimum or maximum value for each parameter required for food safety. A value is just a number. Imagine a dreadful day; there are problems in production. Maybe equipment stalls and product sits. Maybe the electricity flickers and oven temperature drops. Maybe a culture in fermentation isn’t active. Poop happens. What are the values that are absolutely required for the product to be safe? They are often called critical limits. This is the difference between destroying product and selling product. The HACCP plan details the parameters and values required for food safety at each CCP. In production, the operating limits may be different based on quality characteristics or equipment performance, but the product will be safe when critical limits are met. How do you know critical limits are met?
CCPs must be monitored
Every CCP is monitored. Common tools for monitoring are thermometers, timers, flow rate meters, pH probes, and measuring of concentration. Most quality managers want production line monitoring to be automated and continuous. If samples are taken and measured at some frequency, technicians must be trained on the sampling technique, frequency, procedure for measurement and recording of data. The values from monitoring will be compared to critical limits. If the value does not reach the critical limit, the process is out of control and food safety may be compromised. The line operator or technician should be trained to know if the line can be stopped and how to segregate product under question. Depending on the hazard, the product will be evaluated for safety, rerun, released or disposed. When the process is out of control, it is called a deviation from the HACCP plan.
A deviation initiates corrective action and documentation associated with the deviation. You can google examples of corrective action forms; there is no one form required. Basically, the line operator, technician or supervisor starts the paperwork by recording everything about the deviation, evaluation of the product, fate of the product, root cause investigation, and what was done to ensure the problem will not happen again. A supervisor or manager reviews and signs off on the corrective action. The corrective action form and associated documentation should be signed off before the product is released. Sign off is an example of verification. Verification will be discussed in more detail in a future article.
My thoughts on GMPs and HACCP were shared in a webinar on May 2nd hosted by CIJ and NEHA. Please comment on this blog post below. I love feedback!
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
 With markets across the country maturing at a rapid rate, change is a constant. Cannabis companies operating in new markets need to maintain compliance while focusing on their business plan, which can be a difficult task. We sat down with Nick Murer to learn more about compliance issues that cannabis businesses face, like workers comp, payroll taxes, insurance and how outsourcing some HR functions can help.
With markets across the country maturing at a rapid rate, change is a constant. Cannabis companies operating in new markets need to maintain compliance while focusing on their business plan, which can be a difficult task. We sat down with Nick Murer to learn more about compliance issues that cannabis businesses face, like workers comp, payroll taxes, insurance and how outsourcing some HR functions can help. About Nicholas Murer
About Nicholas Murer