On Monday, August 28th, attendees of the Cannabis Science Conference descended on Portland, Oregon for a week of educational talks, networking and studying the science of cannabis. On Monday, Chalice Farms, an extracts and infused products company, hosted the full-day JCanna Boot Camp focused on a deep dive behind the scenes of a cannabis production facility. The Cannabis Science Conference, hosted by Josh Crossney, founder of JCanna, takes place August 28th to 30th.
Attendees touring an extraction setup
Attendees were split into five groups where they listened to a variety of educational sessions and toured the facility. A track focused on cultivation, led by Autumn Karcey, president of Cultivo, Inc., detailed all things facility design for cannabis cultivation, including an in-depth look at sanitation and safety. For example, Karcey discussed HVAC cleanliness, floor-to-ceiling sanitation and the hazards associated with negative pressure. These principles, while applicable to most cultivating facilities, applies particularly to commercial-scale grows in a pharmaceutical setting.
Sandy Mangan and Tristan DeBona demonstrating the grinding technique for sample prep
During one session, Sandy Mangan, accounts manager at SPEX Sample Prep and Tristan DeBona, sales specialist at SPEX Sample Prep, demonstrated the basics of sample preparation for detecting pesticides in infused products, such as gummies. That required using their GenoGrinder and FreezerMill, which uses liquid nitrogen to make gummies brittle, then pulverizing them to a powder-like substance that is more conducive for a QuEChERS preparation.
Joe Konschnik and Susan Steinike demonstrate the QuEChERS method
Joe Konschnik, business development manager at Restek, Susan Steinike, product-marketing manager at Restek and Justin Steimling, an analytical chemist at Restek, gave a demonstration of a full QuEChERS extraction of a cannabis sample for pesticide analysis, with attendees participating to learn the basics of sample preparation for these types of tests.
Following those were some other notable talks, including a tour of the extraction instruments and equipment at Chalice Farms, a look inside their commercial kitchen and a discussion of edibles and product formulation. Dr. Uma Dhanabalan, founder of Uplifting Health and Wellness, a physician with over 30 years of experience in research and patient care, led a discussion of physician participation, patient education and drug delivery mechanisms.
Amanda Rigdon, Emerald Scientific, showing some complex matrices in cannabis products
Amanda Rigdon, chief technical officer of Emerald Scientific, offered a demonstration of easy and adaptable sample preparation techniques for potency testing of infused product matrices. Rigdon showed attendees of the boot camp how wildly diverse cannabis products are and how challenging it can be for labs to test them.
The JCanna Canna Boot Camp is a good example of an educational event catered to the cannabis industry that offers real, hands-on experience and actionable advice. Before the two-day conference this week, the boot camp provided a bird’s eye view for attendees of the science of cannabis.
Terpenes are a group of volatile, unsaturated hydrocarbons found in the essential oils of plants. They are responsible for the characteristic smells and flavors of most plants, such as conifers, citrus, as well as cannabis. Over 140 terpenes have been identified to date and these unique compounds may have medicinal properties. Caryophyllene, for example, emits a sweet, woody, clove taste and is believed to relieve inflammation and produce a neuroprotective effect through CB2 receptor activation. Limonene has a citrus scent and may possess anti-cancer, anti-bacterial, anti-fungal and anti-depression effects. Pinene is responsible for the pine aroma and acts as a bronchodilator. One theory involving terpenes is the Entourage Effect, a synergistic benefit from the combination of cannabinoids and terpenes.
Many customers ask technical service which instrumentation is best, GC or HPLC, for analysis of terpenes. Terpenes are most amenable to GC, due to their inherent volatility. HPLC is generally not recommended; since terpenes have very low UV or MS sensitivity; the cannabinoids (which are present in percent levels) will often interfere or coelute with many of the terpenes.
Figure 1: Terpene profile via headspace, courtesy of ProVerde Laboratories.
Headspace (HS), Solid Phase Microextraction of Headspace (HS-SPME) or Split/Splitless Injection (SSI) are viable techniques and have advantages and disadvantages. While SPME can be performed by either direct immersion with the sample or headspace sampling, HS-SPME is considered the most effective technique since this approach eliminates the complex oil matrix. Likewise, conventional HS also targets volatiles that include the terpenes, leaving the high molecular weight oils and cannabinoids behind (Figure 1). SSI eliminates the complexity of a HS or SPME concentrator/autosampler, however, sensitivity and column lifetime become limiting factors to high throughput, since the entire sample is introduced to the inlet and ultimately the column.
The GC capillary columns range from thicker film, mid-polarity (Rxi-624sil MS for instance) to thinner film, non-polar 100% polysiloxane-based phases, such as an Rxi-1ms. A thicker film provides the best resolution among the highly volatile, early eluting compounds, such as pinene. Heavier molecular weight compounds, such as the cannabinoids, are difficult to bake off of the mid-polarity phases. A thinner, non-polar film enables the heavier terpenes and cannabinoids to elute efficiently and produces sharp peaks. Conversely the early eluting terpenes will often coelute using a thin film column. Columns that do not contain cyano-functional groups (Rxi-624Sil MS), are more robust and have higher temperature limits and lower bleed.
For the GC detector, a Mass Spectrometer (MS) can be used, however, many of the terpenes are isobars, sharing the same ions used for identification and quantification. Selectivity is the best solution, regardless of the detector. The Flame Ionization Detector (FID) is less expensive to purchase and operate and has a greater dynamic range, though it is not as sensitive, nor selective for coeluting impurities.
By accurately and reproducibly quantifying terpenes, cannabis medicines can be better characterized and controlled. Strains, which may exhibit specific medical and psychological traits, can be identified and utilized to their potential. The lab objectives, customer expectations, state regulations, available instrumentation, and qualified lab personnel will ultimately determine how the terpenes will be analyzed.
Edibles and vape pens are rapidly becoming a sizable portion of the cannabis industry as various methods of consumption popularize beyond just smoking dried flower. These products are produced using cannabis concentrates, which come in the form of oils, waxes or shatter (figure 1). Once the cannabinoids and terpenes are removed from the plant material using solvents, the solvent is evaporated leaving behind the product. Extraction solvents are difficult to remove in the low percent range so the final product is tested to ensure leftover solvents are at safe levels. While carbon dioxide and butane are most commonly used, consumer concern over other more toxic residual solvents has led to regulation of acceptable limits. For instance, in Colorado the Department of Public Health and Environment (CDPHE) updated the state’s acceptable limits of residual solvents on January 1st, 2017.
Headspace Analysis
Figure 1: Shatter can be melted and dissolved in a high molecular weight solvent for headspace analysis (HS). Photo Courtesy of Cal-Green Solutions.
Since the most suitable solvents are volatile, these compounds are not amenable to HPLC methods and are best suited to gas chromatography (GC) using a thick stationary phase capable of adequate retention and resolution of butanes from other target compounds. Headspace (HS) is the most common analytical technique for efficiently removing the residual solvents from the complex cannabis extract matrix. Concentrates are weighed out into a headspace vial and are dissolved in a high molecular weight solvent such as dimethylformamide (DMF) or 1,3-dimethyl-3-imidazolidinone (DMI). The sealed headspace vial is heated until a stable equilibrium between the gas phase and the liquid phase occurs inside the vial. One milliliter of gas is transferred from the vial to the gas chromatograph for analysis. Another approach is full evaporation technique (FET), which involves a small amount of sample sealed in a headspace vial creating a single-phase gas system. More work is required to validate this technique as a quantitative method.
Gas Chromatographic Detectors
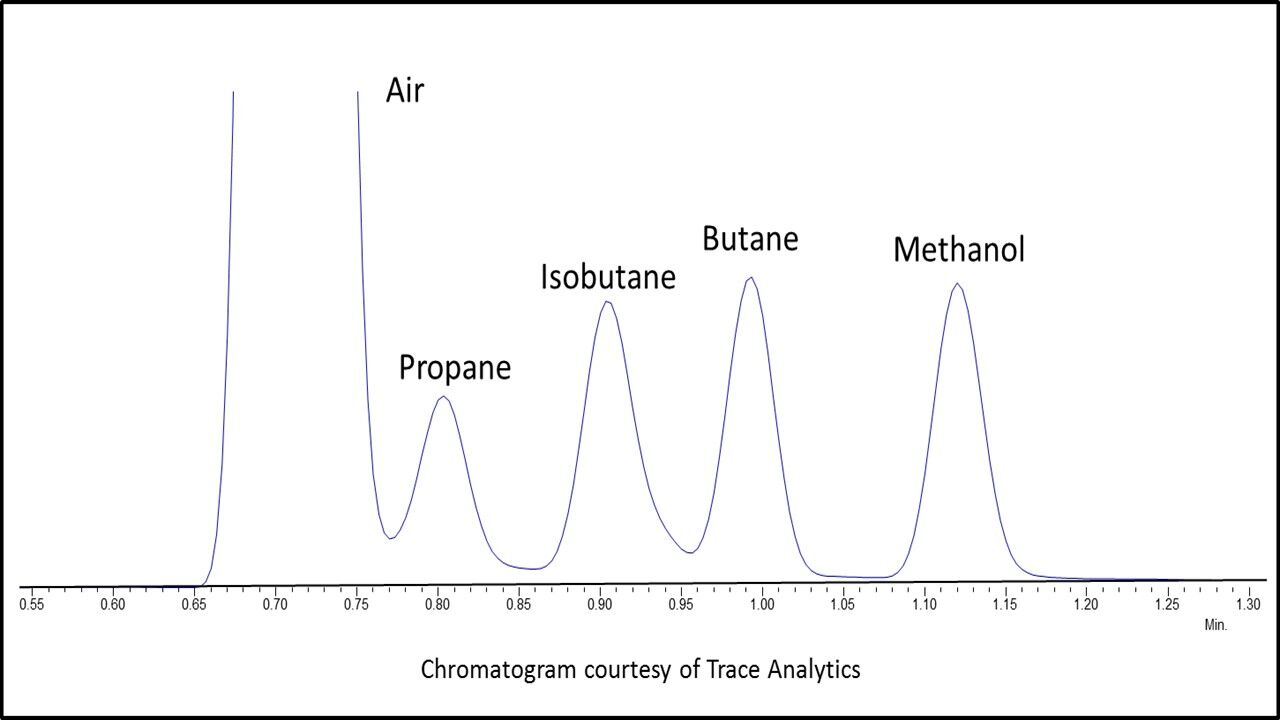
The flame ionization detector (FID) is selective because it only responds to materials that ionize in an air/hydrogen flame, however, this condition covers a broad range of compounds. When an organic compound enters the flame; the large increase in ions produced is measured as a positive signal. Since the response is proportional to the number of carbon atoms introduced into the flame, an FID is considered a quantitative counter of carbon atoms burned. There are a variety of advantages to using this detector such as, ease of use, stability, and the largest linear dynamic range of the commonly available GC detectors. The FID covers a calibration of nearly 5 orders of magnitude. FIDs are inexpensive to purchase and to operate. Maintenance is generally no more complex than changing jets and ensuring proper gas flows to the detector. Because of the stability of this detector internal standards are not required and sensitivity is adequate for meeting the acceptable reporting limits. However, FID is unable to confirm compounds and identification is only based on retention time. Early eluting analytes have a higher probability of interferences from matrix (Figure 2).
Figure 2: Resolution of early eluting compounds by headspace – flame ionization detection (HS-FID). Chromatogram Courtesy of Trace Analytics.
Mass Spectrometry (MS) provides unique spectral information for accurately identifying components eluting from the capillary column. As a compound exits the column it collides with high-energy electrons destabilizing the valence shell electrons of the analyte and it is broken into structurally significant charged fragments. These fragments are separated by their mass-to-charge ratios in the analyzer to produce a spectral pattern unique to the compound. To confirm the identity of the compound the spectral fingerprint is matched to a library of known spectra. Using the spectral patterns the appropriate masses for quantification can be chosen. Compounds with higher molecular weight fragments are easier to detect and identify for instance benzene (m/z 78), toluene (m/z 91) and the xylenes (m/z 106), whereas low mass fragments such as propane (m/z 29), methanol (m/z 31) and butane (m/z 43) are more difficult and may elute with matrix that matches these ions. Several disadvantages of mass spectrometers are the cost of equipment, cost to operate and complexity. In addition, these detectors are less stable and require an internal standard and have a limited dynamic range, which can lead to compound saturation.
Regardless of your method of detection, optimized HS and GC conditions are essential to properly resolve your target analytes and achieve the required detection limits. While MS may differentiate overlapping peaks the chances of interference of low molecular weight fragments necessitates resolution of target analytes chromatographically. FID requires excellent resolution for accurate identification and quantification.
As mentioned in Part 1, pesticides residue analysis is very challenging especially considering the complexity of cannabis and the variety of flower, concentrates and infused products. In addition, pesticides are tested at low levels typically at parts-per-billion (ppb). For example, the food safety industry often uses 10 ppb as a benchmark limit of quantification. To put that in perspective, current pesticides limits in cannabis range from 10 ppb default (Massachusetts Regulatory Limit) to a more typical range of 100 ppb to 2 ppm in other states. Current testing is also complicated by evolving regulations.
Despite these challenges, adaptation of methods used by the food safety industry have proved successful for testing pesticides in cannabis. These methods typically rely on mass spectrometric detection paired with sample preparation methods to render the sample clean enough to yield quality data.
Pesticide Analysis Methods: Sample preparation and Analytical Technique Strategy
Generally, methods can be divided into two parts; sample preparation and analytical testing where both are critical to the success of pesticide residue testing and are inextricably linked. Reliance on mass spectrometric techniques like tandem mass spectrometry and high resolution accurate mass (HRAM) mass spectrometry is attributed to the substantial sensitivity and selectivity provided. The sensitivity and selectivity achievable by the detector largely dictates the sample preparation that will be required. The more sensitive and selective the detector, the less rigorous and resource intensive sample preparation can be.
Analytical technique: Gas and Liquid Chromatography Tandem Mass Spectrometry
The workhorse approach for pesticide residue analysis involves using gas chromatography and liquid chromatography tandem mass spectrometry (MS/MS) in the ion transition mode. This ion transition mode, often referred to as multiple reaction monitoring (MRM) or selected reaction monitoring (SRM), adds the selectivity and sensitivity needed for trace level analysis. Essentially, a pesticide precursor ion is fragmented into product ions. The detector monitors the signal for a specified product ion known to have originated from the pesticide precursor ion. This allows the signal to be corrected, associated with the analyte and not with other matrix components in the sample. In addition, because only ions meeting the precursor/product ion requirements are passed to the detector with little noise, there is a benefit to the observed signal to noise ratio allowing better sensitivity than in other modes. Even though ion transitions are specific, there is the possibility a matrix interference that also demonstrates that same ion transition could result in a false positive. Multiple ion transitions for each analyte are monitored to determine an ion ratio. The ion ratio should remain consistent for a specific analyte and is used to add confidence to analyte identification.
The best choice for pesticide analysis between gas chromatography (GC) and liquid chromatography (LC) is often questioned. To perform comprehensive pesticide screening similar to the way the food safety market approaches this challenge requires both techniques. It is not uncommon for screening methods to test for several hundred pesticides that vary in physiochemical properties. It may be possible that with a smaller list of analytes, only one technique will be needed but often in order to reach the low limits for pesticide residues both GC and LC are required.
Modified QuEChERS extraction using 1.5 grams of cannabis flower. Courtesy of Julie Kowalski (Restek Corporation), Jeff Dahl (Shimadzu Scientific Instruments) and Derek Laine (Trace Analytics).
Analytical technique: Sample Preparation
Less extensive sample preparation is possible when combined with sensitive and selective detectors like MS/MS. One popular method is the QuEChERS approach. QuEChERS stands for Quick, Easy, Cheap, Effective, Rugged and Safe. It consists of a solvent extraction/salting out step followed by a cleanup using dispersive solid phase extraction. Originally designed for fruit and vegetable pesticide testing, QuEChERS has been modified and used for many other commodity types including cannabis. Although QuEChERS is a viable method, sometimes more cleanup is needed and this can be done with cartridge solid phase extraction. This cleanup functions differently and is more labor intensive, but results in a cleaner extract. A cleaner extract helps to secure quality data and is sometimes needed for difficult analyses.
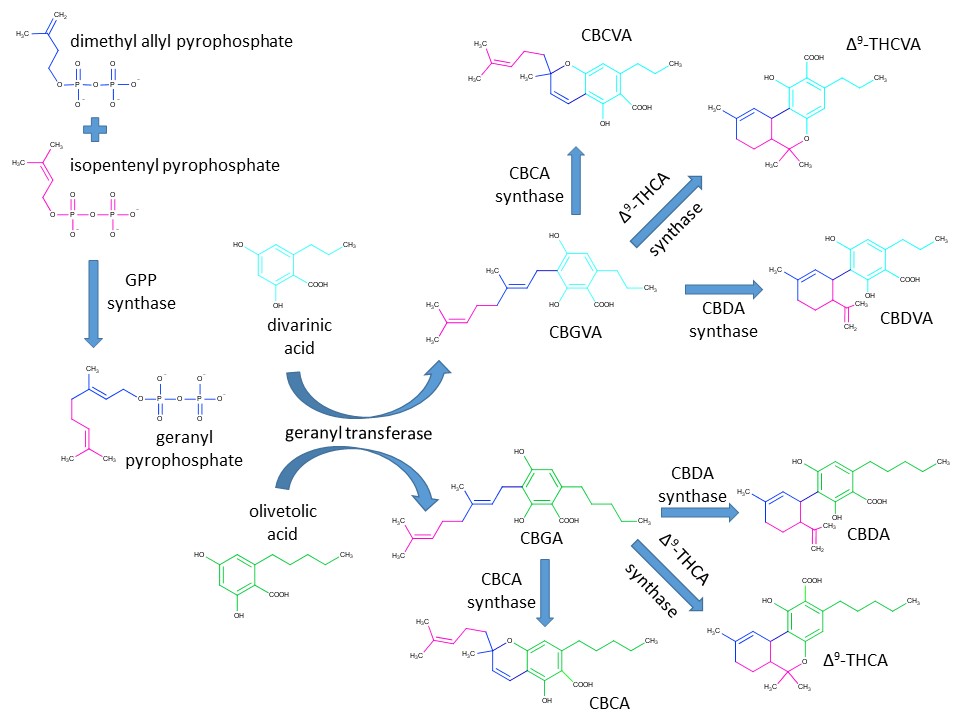
As mentioned in Part 1, the physiological effects of cannabis are mediated by a group of structurally related organic compounds known as cannabinoids. The cannabinoids are biosynthetically produced by a growing cannabis plant and Figure 1 details the biosynthetic pathways leading to some of the most important cannabinoids in plant material.
Figure 1: The biosynthetic pathway of phytocannabinoid production in cannabis has been deeply studied through isotopic labeling experiments
The analytical measurement of cannabinoids is important to ensure the safety and quality of cannabis as well as its extracts and edible formulations. Total cannabinoid levels can vary significantly between different cultivars and batches, from about 5% up to 20% or more by dry weight. Information on cannabinoid profiles can be used to tailor cultivars for specific effects and allows end users to select an appropriate dose.
Routine Analysisvs. Cannabinomics
Several structurally analogous groups of cannabinoids exist. In total, structures have been assigned for more than 70 unique phytocannabinoids as of 2005 and the burgeoning field of cannabinomics seeks to comprehensively measure these compounds.¹
Considering practical potency analysis, the vast majority of cannabinoid content is accounted for by 10-12 compounds. These include Δ9-tetrahydrocannabinol (THC), cannabidiol (CBD), cannabigerol (CBG), Δ9-tetrahydrocannabivarian (THCV), cannabidivarin (CBDV) and their respective carboxylic acid forms. The cannabinoids occur primarily as carboxylic acids in plant material. Decarboxylation occurs when heat is applied through smoking, vaporization or cooking thereby producing neutral cannabinoids which are more physiologically active.
Potency Analysis by HPLC and GC
Currently, HPLC and GC are the two most commonly used techniques for potency analysis. In the case of GC, the heat used to vaporize the injected sample causes decarboxylation of the native cannabinoid acids. Derivatization of the acids may help reduce decarboxylation but overall this adds another layer of complexity to the analysis² ³. HPLC is the method of choice for direct analysis of cannabinoid profiles and this technique will be discussed further.
A sample preparation method consisting of grinding/homogenization and alcohol extraction is commonly used for cannabis flower and extracts. It has been shown to provide good recovery and precision² ³. An aliquot of the resulting extract can then be diluted with an HPLC compatible solvent such as 25% water / 75% acetonitrile with 0.1% formic acid. The cannabinoids are not particularly water soluble and can precipitate if the aqueous percentage is too high.
To avoid peak distortion and shifting retention times the diluent and initial mobile phase composition should be reasonably well matched. Another approach is to make a smaller injection (1-2 µL) of a more dissimilar solvent. The addition of formic acid or ammonium formate buffer acidifies the mobile phase and keeps the cannabinoid acids protonated.
The protonated acids are neutral and thus well retained on a C18 type column, even at higher (~50% or greater) concentrations of organic solvent² ³.
Detection is most often done using UV absorbance. Two main types of UV detectors are available for HPLC, single wavelength and diode array. A diode array detector (DAD) measures absorbance across a range of wavelengths producing a spectrum at each point in a chromatogram while single wavelength detectors only monitor absorbance at a single user selected wavelength. The DAD is more expensive, but very useful for detecting coelutions and interferences.
References
Chemical Constituents of Marijuana: The Complex Mixture of Natural Cannabinoids. Life Sciences, 78, (2005), pp. 539
Development and Validation of a Reliable and Robust Method for the Analysis of Cannabinoids and Terpenes in Cannabis. Journal of AOAC International, 98, (2015), pp. 1503
Innovative Development and Validation of an HPLC/DAD Method for the Qualitative and Quantitative Determination of Major Cannabinoids in Cannabis Plant Material. Journal of Chromatography B, 877, (2009), pp. 4115
Rebecca is an Applications Scientist at Restek Corporation and is eager to field any questions or comments on cannabis analysis, she can be reached by e-mail, rebecca.stevens@restek.com or by phone at 814-353-1300 (ext. 2154)
In the last article I referred to the analogy of the analytical reference material being a keystone of the laboratory foundation, the stone upon which all data relies. I then described the types of reference materials and their use in analytical testing in general terms. This article will describe the steps required to properly manufacture and deliver a certified reference material (CRM) along with the necessary documentation.
A CRM is an exclusive reference material that meets strict criteria defined by ISO Guide 34 and ISO/IEC 17025. ISO is the International Organization for Standardization and IEC is the International Electrotechnical Commission. These organizations work together to set globally recognized standards. In order for a reference material to be labeled as a CRM it must 1) be made with raw or starting materials which are characterized using qualified methods and instruments, 2) be produced in an ISO-accredited lab under documented procedures, and 3) fall under the manufacturer’s scopes of accreditation. Verifying a CRM supplier has these credentials is easily done by viewing their certificates which should include their scopes of accreditation.
There are many steps required to produce a CRM that meets the above three criteria. The first step requires a review of the customer’s, or end-user’s requirements to carefully define what is to be tested, at what levels and which analytical workflow will be used. Such information enables the producer to identify the proper compounds and solvents required to properly formulate the requested CRM.
The next step requires sourcing and acquiring the raw, or starting materials, then verifying their compatibility and stability using stability and shipping studies in accordance with ISO requirements. Next the chemical identify and purity of the raw materials must be characterized using one or more analytical techniques such as: GC-FID, HPLC, GC-ECD, GC-MS, LC-MS, refractive index and melting point. In some cases, the percent purity is changed by the producer when their testing verifies it’s different from the supplier label. All steps are of course documented.
The producer’s analytical balances must be verified using NIST traceable weights and calibrated annually by an accredited third party provider to guarantee accurate measurement. CRMs must be prepared using Class A volumetric glassware, and all ampules and vials used in preparation and final packaging must be chemically treated to prevent compound degradation during storage. Next, CRMs are packaged in an appropriate container, labeled then properly stored to maintain the quality and stability until it’s ready to be shipped. All labels must include critical storage, safety and shelf life information to meet federal requirements. The label information must be properly linked to documentation commonly referred to as a certificate of analysis (COA) which describes all of the above steps and verifies the traceability and uncertainty of all measurements for each compound contained in the CRM.
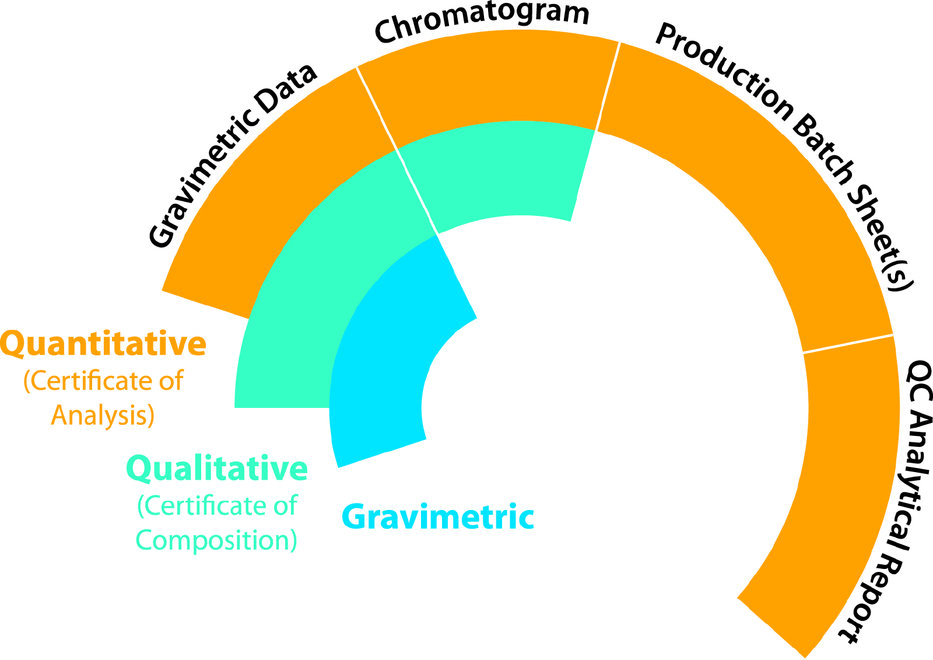
My company, RESTEK, offers a variety of documentation choices to accompany each CRM. Depending on the intended use and data quality objectives specified by the end-user, which were defined way back at the first step, three options are typically offered: They include gravimetric only, qualitative which includes gravimetric, and fully quantitative which includes all three levels of documentation. The graphic to the right summarizes the three options and what they include.
It’s important to understand which level you’re purchasing especially when ordering a custom CRM from a supplier. Most stock CRMs include all three levels of documentation, but it’s important to be sure.
Understanding what must be done to produce and deliver a CRM sets it apart from other reference material types, however it’s important to understand there are some instances where CRMs are either not available, nor required and in those situations other types of reference materials are perfectly acceptable.
If you have any questions or would like more details about reference materials please contact me, Joe Konschnik at (800) 356-1688 ext. 2002 by phone, or email me at joe.konschnik@restek.com.
In previous articles, you may recall that Amanda Rigdon, one our contributing authors, stated that instrument calibration is the foundation of all data quality. In this article, I would like to expand on that salient point. A properly calibrated instrument will, in fact, produce reliable data. It is the foundation we build our data upon. All foundations are comprised of building blocks, and our laboratory is no exception. If we take this analogy further, the keystone to the laboratory foundation, the stone that all data relies upon, is the analytical reference material. Proper calibration means that it is based on a true, accurate value. That is what the reference material provides. In this article, I would like to expand on the use and types of reference materials in analytical testing.
To develop sound analytical data, it is important to understand the significance of reference materials and how they are properly used. The proper selection and use of reference materials ensures the analytical certainty, traceability and comparability necessary to produce scientifically sound data. First, let’s take a moment to define the types of commonly used reference materials. According to the International Vocabulary of Metrology (VIM), a Reference Standard (RS) is something that is reused to measure against, like a balance or a set of weights. A Reference Material (RM) is a generic term. It is described as something that is prepared using a RS that is homogeneous, stable and is consumed during its use for measurement. An example of an RM is the solutions used to construct a calibration curve, often referred to as calibration standards, on your GC or LC. Due to the current state of cannabis testing, reference materials can be hard to find and, even more critical, variable in their accuracy to a known reference standard. Sometimes this is not critical, but when quantifying an unknown, it is paramount.
RMs can be either quantitative or qualitative. Qualitative RMs verify the identity and purity of a compound. Quantitative RMs, on the other hand, provide a known concentration, or mass, telling us not only what is present, and its purity, but also how much. This is typically documented on the certificate that accompanies the reference material, which is provided by the producer or manufacturer. The certificate describes all of the properties of the starting materials and steps taken to prepare the RM. For testing requirements, like potency, pesticides, etc., where quantitation is expected, it is important to use properly certified quantitative RMs.
Now, the pinnacle of reference materials is the Certified Reference Material (CRM). VIM defines a Certified Reference Material (CRM) as an RM accompanied by documentation issued by an authoritative body and provides one or more specified property values, with associated uncertainties and traceability using valid procedures. A CRM is generally recognized as providing the highest level of traceability and accuracy to a measurement – the strongest keystone you can get for your foundation. It is also important to recognize that the existence of a certificate does not make a reference material a CRM. It is the process used in manufacturing that makes it a CRM, and these are typically accreditations earned by specific manufacturers who have invested on this level of detail.
Now that we understand the types of reference materials we can choose, in the next article of this series we will describe what a CRM provider must do to ensure the material and how we can use them to develop reliable data. Without properly formulated and prepared CRMs, instrument calibration and the use of internal standards are less effective at ensuring the quality of your data.
If you have any questions please contact me, Joe Konschnik at (800) 356-1688 ext. 2002 by phone, or email me at joe.konschnik@restek.com.
Emerald Scientific, a supplier of reagents, supplies, equipment and services to cannabis testing and extraction facilities, recently named Amanda Rigdon as the company’s chief technology officer. Rigdon previously worked at Restek Corporation, a manufacturer of chromatography supplies, as an applications chemist and a member of their gas chromatography columns product marketing team.
Amanda Rigdon, chief technology officer at Emerald Scientific
Before working in the cannabis space, Rigdon began her career in the pharmaceutical and clinical/forensics industries. She spent seven years in Restek’s applications lab where she was responsible for the development and application of chromatography products for the pharmaceutical and clinical/forensics arenas. In recent years, she has been an outspoken advocate in the science of cannabis while with Restek.
As a strong proponent for scientific progress in cannabis, she brings extensive technical expertise and marketing experience related to cannabis testing and research. Presenting at numerous cannabis science conferences and seminars, she regularly provides education on analytical methods and best practices in the lab.
As a contributing author to CannabisIndustryJournal.com and member of the editorial advisory board, she writes a column addressing challenges in the lab and providing technical advice. “I’m thrilled to be a part of the Emerald Scientific team and a member of the cannabis community as a whole,” says Rigdon. “I’ve known the folks at Emerald [Scientific] for years; they’re among the best in the business, and they’ve been supporting the cannabis community since the early days of cannabis analytics.” Rigdon’s mantra in the cannabis testing space has long been to support sound science in the interest of protecting patient and consumer health.
“I’m really looking forward to using my technical skills in conjunction with Emerald’s position and reach in the market to make work easier for cannabis labs through education, applications and new products,” adds Rigdon. Emerald Scientific is widely known in the cannabis testing community for The Emerald Test, an inter- laboratory comparison proficiency test, organized twice per year. It also hosts The Emerald Conference, an annual scientific meeting for scientists, policy makers, producers, and other key members of the cannabis industry. ˇThe Emerald Conference is the first scientifically focused conference for the cannabis industry, now coming up on its third annual conference in February 2017.
Everyone likes to have a safety net, and scientists are no different. This month I will be discussing internal standards and how we can use them not only to improve the quality of our data, but also give us some ‘wiggle room’ when it comes to variation in sample preparation. Internal standards are widely used in every type of chromatographic analysis, so it is not surprising that their use also applies to common cannabis analyses. In my last article, I wrapped up our discussion of calibration and why it is absolutely necessary for generating valid data. If our calibration is not valid, then the label information that the cannabis consumer sees will not be valid either. These consumers are making decisions based on that data, and for the medical cannabis patient, valid data is absolutely critical. Internal standards work with calibration curves to further improve data quality, and luckily it is very easy to use them.
So what are internal standards? In a nutshell, they are non-analyte compounds used to compensate for method variations. An internal standard can be added either at the very beginning of our process to compensate for variations in sample prep and instrument variation, or at the very end to compensate only for instrument variation. Internal standards are also called ‘surrogates’, in some cases, however, for the purposes of this article, I will simply use the term ‘internal standard.’
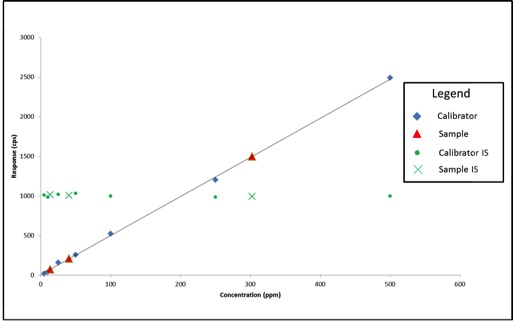
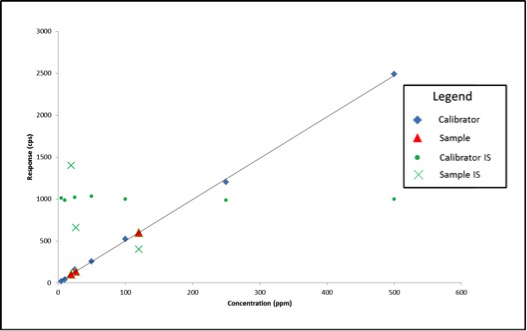
Now that we know what internal standards are, lets look at how to use them. We use an internal standard by adding it to all samples, blanks, and calibrators at the same known concentration. By doing this, we now have a single reference concentration for all response values produced by our instrument. We can use this reference concentration to normalize variations in sample preparation and instrument response. This becomes very important for cannabis pesticide analyses that involve lots of sample prep and MS detectors. Figure 1 shows a calibration curve plotted as we saw in the last article (blue diamonds), as well as the response for an internal standard added to each calibrator at a level of 200ppm (green circles). Additionally, we have three sample results (red triangles) plotted against the calibration curve with their own internal standard responses (green Xs).
Figure 1: Calibration Curve with Internal Standard Responses and Three Sample Results
In this case, our calibration curve is beautiful and passes all of the criteria we discussed in the previous article. Lets assume that the results we calculate for our samples are valid – 41ppm, 303ppm, and 14ppm. Additionally, we can see that the responses for our internal standards make a flat line across the calibration range because they are present at the same concentration in each sample and calibrator. This illustrates what to expect when all of our calibrators and samples were prepared correctly and the instrument performed as expected. But lets assume we’re having one of those days where everything goes wrong, such as:
We unknowingly added only half the volume required for cleanup for one of the samples
The autosampler on the instrument was having problems and injected the incorrect amount for the other two samples
Figure 2 shows what our data would look like on our bad day.
Figure 2: Calibration Curve with Internal Standard Responses and Three Sample Results after Method Errors
We experienced no problems with our calibration curve (which is common when using solvent standard curves), therefore based on what we’ve learned so far, we would simply move on and calculate our sample results. The sample results this time are quite different: 26ppm, 120ppm, and 19ppm. What if these results are for a pesticide with a regulatory cutoff of 200ppm? When measured accurately, the concentration of sample 2 is 303ppm. In this example, we may have unknowingly passed a contaminated product on to consumers.
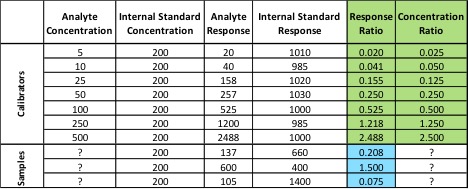
In the first two examples, we haven’t been using our internal standard – we’ve only been plotting its response. In order to use the internal standard, we need to change our calibration method. Instead of plotting the response of our analyte of interest versus its concentration, we plot our response ratio (analyte response/internal standard response) versus our concentration ratio (analyte concentration/internal standard concentration). Table 1 shows the analyte and internal standard response values for our calibrators and samples from Figure 2.
Table 1: Values for Calibration Curve and Samples Using Internal Standard
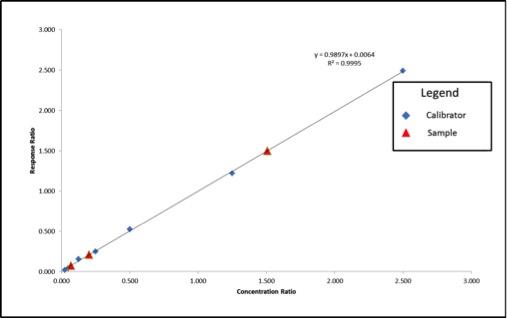
The values highlighted in green are what we will use to build our calibration curve, and the values in blue are what we will use to calculate our sample concentration. Figure 3 shows what the resulting calibration curve and sample points will look like using an internal standard.
Figure 3: Calibration Curve and Sample Results Calculated Using Internal Standard Correction
We can see that our axes have changed for our calibration curve, so the results that we calculate from the curve will be in terms of concentration ratio. We calculate these results the same way we did in the previous article, but instead of concentrations, we end up with concentration ratios. To calculate the sample concentration, simply multiply by the internal standard amount (200ppm). Figure 4 shows an example calculation for our lowest concentration sample.
Figure 4: Example Calculation for Sample Results for Internal-Standard Corrected Curve
Using the calculation shown in Figure 4, our sample results come out to be 41ppm, 302ppm, and 14ppm, which are accurate based on the example in Figure 1. Our internal standards have corrected the variation in our method because they are subjected to that same variation.
As always, there’s a lot more I can talk about on this topic, but I hope this was a good introduction to the use of internal standards. I’ve listed couple of resources below with some good information on the use of internal standards. If you have any questions on this topic, please feel free to contact me at amanda.rigdon@restek.com.
Despite the title, this article is not about weight loss – it is about generating valid analytical data for quantitative analyses. In the last installment of The Practical Chemist, I introduced instrument calibration and covered a few ways we can calibrate our instruments. Just because we have run several standards across a range of concentrations and plotted a curve using the resulting data, it does not mean our curve accurately represents our instrument’s response across that concentration range. In order to be able to claim that our calibration curve accurately represents our instrument response, we have to take a look at a couple of quality indicators for our curve data:
correlation coefficient (r) or coefficient of determination (r2)
back-calculated accuracy (reported as % error)
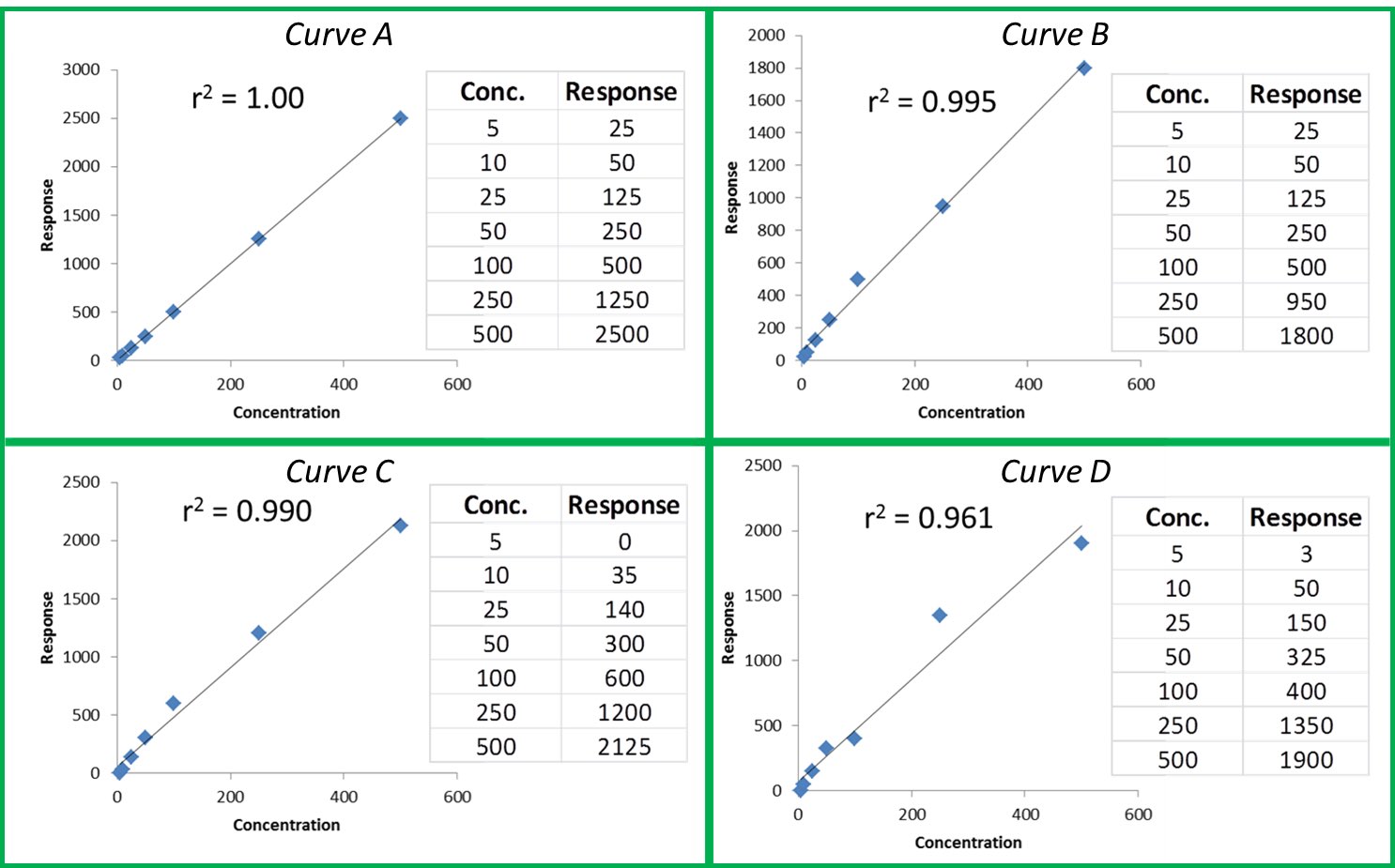
The r or r2 values that accompany our calibration curve are measurements of how closely our curve matches the data we have generated. The closer the values are to 1.00, the more accurately our curve represents our detector response. Generally, r values ≥0.995 and r2 values ≥ 0.990 are considered ‘good’. Figure 1 shows a few representative curves, their associated data, and r2 values (concentration and response units are arbitrary).
Figure 1: Representative Curves and r2 values
Let’s take a closer look at these curves:
Curve A: This represents a case where the curve perfectly matches the instrument data, meaning our calculated unknown values will be accurate across the entire calibration range.
Curve B: The r2 value is good and visually the curve matches most of the data points pretty well. However, if we look at our two highest calibration points, we can see that they do not match the trend for the rest of the data; the response values should be closer to 1250 and 2500. The fact that they are much lower than they should be could indicate that we are starting to overload our detector at higher calibration levels; we are putting more mass of analyte into the detector than it can reliably detect. This is a common problem when dealing with concentrated samples, so it can occur especially for potency analyses.
Curve C: We can see that although our r2 value is still okay, we are not detecting analytes as we should at the low end of our curve. In fact, at our lowest calibration level, the instrument is not detecting anything at all (0 response at the lowest point). This is a common problem with residual solvent and pesticide analyses where detection levels for some compounds like benzene are very low.
Curve D: It is a perfect example of our curve not representing our instrument response at all. A curve like this indicates a possible problem with the instrument or sample preparation.
So even if our curve looks good, we could be generating inaccurate results for some samples. This brings us to another measure of curve fitness: back-calculated accuracy (expressed as % error). This is an easy way to determine how accurate your results will be without performing a single additional run.
Back-calculated accuracy simply plugs the area values we obtained from our calibrators back into the calibration curve to see how well our curve will calculate these values in relation to the known value. We can do this by reprocessing our calibrators as unknowns or by hand. As an example, let’s back-calculate the concentration of our 500 level calibrator from Curve B. The formula for that curve is: y = 3.543x + 52.805. If we plug 1800 in for y and solve for x, we end up with a calculated concentration of 493. To calculate the error of our calculated value versus the true value, we can use the equation: % Error = [(calculated value – true value)/true value] * 100. This gives us a % error of -1.4%. Acceptable % error values are usually ±15 – 20% depending on analysis type. Let’s see what the % error values are for the curves shown in Figure 1.
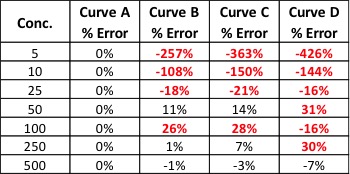
Table 1: % Error for Back-Calculated Values for Curves A – D
Our % error values have told us what our r2 values could not. We knew Curve D was unacceptable, but now we can see that Curves B and C will yield inaccurate results for all but the highest levels of analyte – even though the results were skewed at opposite ends of the curves.
There are many more details regarding generating calibration curves and measuring their quality that I did not have room to mention here. Hopefully, these two articles have given you some tools to use in your lab to quickly and easily improve the quality of your data. If you would like to learn more about this topic or have any questions, please don’t hesitate to contact me at amanda.rigdon@restek.com.
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.